
Anti-Colonization Effect of Au Surfaces with Self-Assembled Molecular Monolayers Functionalized with Antimicrobial Peptides on S. epidermidis

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Surface Design and Preparation
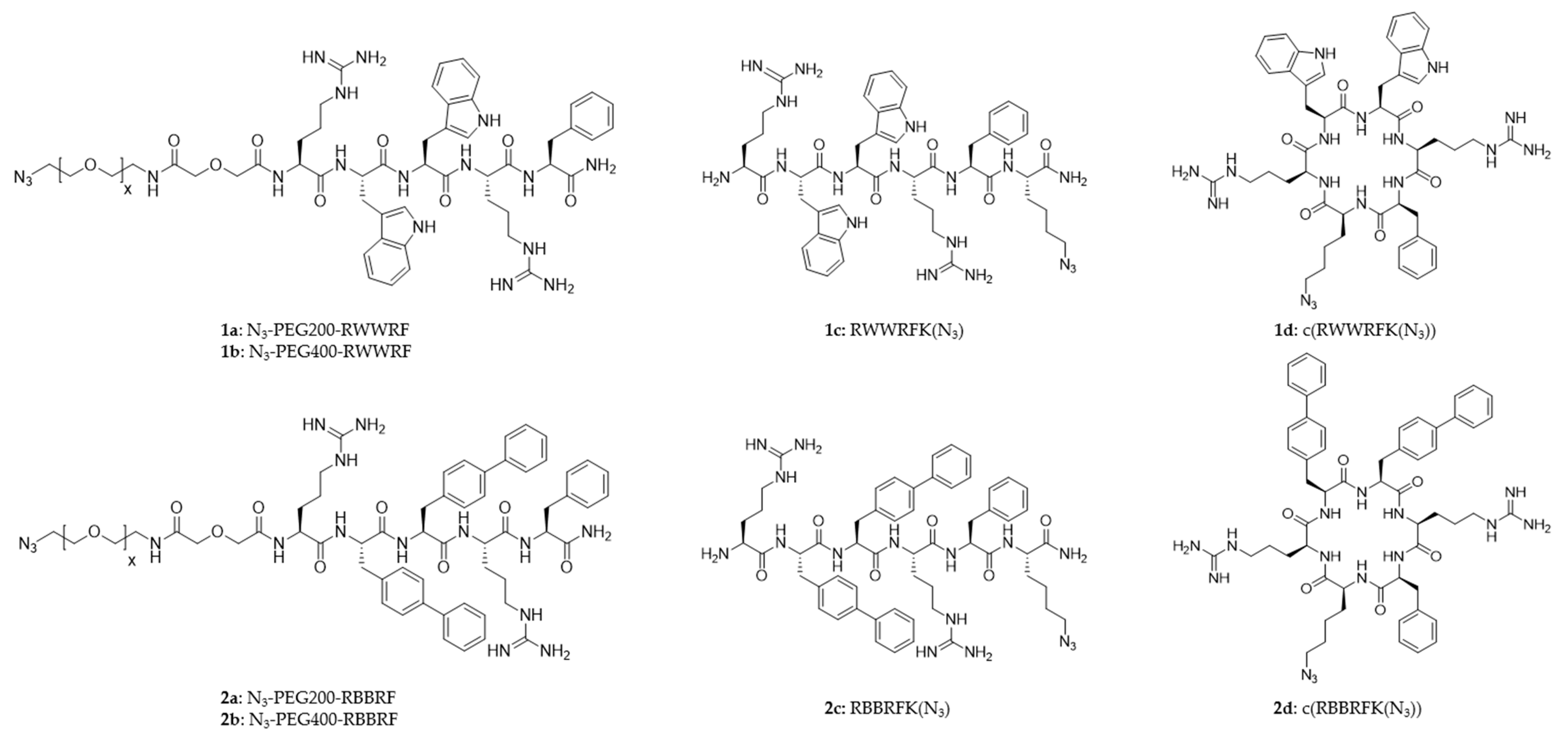
2.2. Peptide Design
2.3. Intrinsic Antimicrobial Activity of the Peptides
2.4. Characterization of Peptide Surfaces
2.4.1. Contact Angle
2.4.2. Surface Characterization by Spatially Resolved ToF-SIMS Mass Spectrometry
2.5. Anti-Colonization Efficacy of Peptide Modified Gold Surfaces
Certika
3. Discussion
3.1. The Intrinsic Antimicrobial Activity of the Peptide Library
3.2. Surface Attachment of the Peptides
3.2.1. Contact Angle and Surface Lipophilicity
3.2.2. Verification of Surface Integrity and Homogeneity by Spatially Resolved ToF-SIMS Mass Spectrometry
3.3. Anti-Colonization Efficacy
4. Materials and Methods
4.1. Materials
4.2. Experimental Method
4.2.1. Synthesis of Azide and Carboxylic Acid Terminal-Conjugated Polyethylene Glycol
4.2.2. Synthesis of Linear Azidopeptides and Azido PEG Peptides
4.2.3. Synthesis of Cyclic Azidopeptides
4.2.4. HPLC
4.2.5. Preparation of Au Surface and Copper(I)-Catalyzed Alkyne-Azide Cycloaddition
4.2.6. Minimal Inhibitory Concentration Determinations
4.2.7. Certika Assay
4.2.8. ToF-SIMS Mass Spectrometry Imaging
4.2.9. Contact Angle Measurements
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Health Care-Associated Infections FACT SHEET. Available online: https://www.who.int/gpsc/country_work/gpsc_ccisc_fact_sheet_en.pdf (accessed on 28 November 2021).
- Centers for Disease Control and Prevention. Types of Healthcare-Associated Infections; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014. Available online: https://www.cdc.gov/hai/infectiontypes.html (accessed on 28 November 2021).
- U.S. Department of Health and Human Services. National HAI Targets & Metrics; U.S. Department of Health and Human Services: Washington, DC, USA, 2020. Available online: https://health.gov/our-work/health-care-quality/health-care-associated-infections/targets-metrics (accessed on 28 November 2021).
- U.S. Department of Health and Human Services. National Action Plan to Prevent Health Care-Associated Infections: Road Map to Elimination; U.S. Department of Health and Human Services: Washington, DC, USA, 2020. Available online: https://health.gov/our-work/health-care-quality/health-care-associated-infections/national-hai-action-plan (accessed on 1 December 2021).
- Chandki, R.; Banthia, P.; Banthia, R. Biofilms: A microbial home. J. Indian Soc. Periodontol. 2011, 15, 111–114. [Google Scholar]
- Donlan, R.M. Biofilms and device-associated infections. Emerg. Infect. Dis. 2001, 7, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Haddadin, Y.; Annamaraju, P.; Regunath, H. Central Line Associated Blood Stream Infections (CLABSI); StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Weiner-Lastinger, L.M.; Abner, S.; Edwards, J.R.; Kallen, A.J.; Karlsson, M.; Magill, S.S.; Pollock, D.; See, I.; Soe, M.M.; Walters, M.S.; et al. Antimicrobial-resistant pathogens associated with adult healthcare-associated infections: Summary of data reported to the National Healthcare Safety Network, 2015–2017. Infect. Control. Hosp. Epidemiol. 2019, 41, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Girona-Alarcón, M.; Fresán, E.; Garcia-Garcia, A.; Bobillo-Perez, S.; Balaguer, M.; Felipe, A.; Esteban, M.E.; Jordan, I. Device-associated multidrug-resistant bacteria surveillance in critically ill children: 10 years of experience. Acta Paediatr. 2020, 110, 203–209. [Google Scholar] [CrossRef]
- Rupp, M.E.; Fitzgerald, T.; Marion, N.; Helget, V.; Puumala, S.; Anderson, J.R.; Fey, P.D. Effect of silver-coated urinary catheters: Efficacy, cost-effectiveness, and antimicrobial resistance. Am. J. Infect. Control. 2004, 32, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Montero, D.A.; Arellano, C.; Pardo, M.; Vera, R.; Gálvez, R.; Cifuentes, M.; Berasain, M.A.; Gómez, M.; Ramírez, C.; Vidal, R.M. Antimicrobial properties of a novel copper-based composite coating with potential for use in healthcare facilities. Antimicrob. Resist. Infect. Control 2019, 8, 3. [Google Scholar] [CrossRef]
- Choi, Y.J.; Lim, J.K.; Park, J.J.; Huh, H.; Kim, D.-J.; Gong, C.-H.; Yoon, S.Z. Chlorhexidine and silver sulfadiazine coating on central venous catheters is not sufficient for protection against catheter-related infection: Simulation-based laboratory research with clinical validation. J. Int. Med. Res. 2017, 45, 1042–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dave, R.N.; Joshi, H.; Venugopalan, V.P. Novel Biocatalytic Polymer-Based Antimicrobial Coatings as Potential Ureteral Biomaterial: Preparation andIn VitroPerformance Evaluation. Antimicrob. Agents Chemother. 2010, 55, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Trop, M.; Novak, M.; Rodl, S.; Hellbom, B.; Kroell, W.; Goessler, W. Silver-Coated Dressing Acticoat Caused Raised Liver Enzymes and Argyria-like Symptoms in Burn Patient. J. Trauma Inj. Infect. Crit. Care 2006, 60, 648–652. [Google Scholar] [CrossRef]
- Wan, A.T.; Conyers, R.A.; Coombs, C.J.; Masterton, J.P. Determination of silver in blood, urine, and tissues of volunteers and burn patients. Clin. Chem. 1991, 37, 1683–1687. [Google Scholar] [CrossRef] [PubMed]
- Wesgate, R.; Grasha, P.; Maillard, J.-Y. Use of a predictive protocol to measure the antimicrobial resistance risks associated with biocidal product usage. Am. J. Infect. Control 2016, 44, 458–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdankhah, S.P.; Scheie, A.A.; Høiby, E.A.; Lunestad, B.-T.; Heir, E.; Fotland, T.; Naterstad, K.; Kruse, H. Triclosan and Antimicrobial Resistance in Bacteria: An Overview. Microb. Drug Resist. 2006, 12, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Chuanchuen, R.; Beinlich, K.; Hoang, T.T.; Becher, A.; Karkhoff-Schweizer, R.R.; Schweizer, H.P. Cross-resistance between triclosan and antibiotics in Pseudomonas aeruginosa is mediated by multidrug efflux pumps: Exposure of a susceptible mutant strain to triclosan selects nfxB mutants overexpressing MexCD-OprJ. Antimicrob. Agents Chemother. 2001, 45, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, F.; Carvalho, I.F.; Montelaro, R.C.; Gomes, P.; Martins, M.C.L. Covalent immobilization of antimicrobial peptides (AMPs) onto biomaterial surfaces. Acta Biomater. 2011, 7, 1431–1440. [Google Scholar] [CrossRef] [Green Version]
- Riool, M.; De Breij, A.; Drijfhout, J.W.; Nibbering, P.H.; Zaat, S.A.J. Antimicrobial Peptides in Biomedical Device Manufacturing. Front. Chem. 2017, 5, 63. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Fjell, C.; Hiss, J.A.; Hancock, R.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Pirri, G.; Nicoletto, S. Antimicrobial peptides: An overview of a promising class of therapeutics. Open Life Sci. 2007, 2, 1–33. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Raheem, N.; Straus, S.K. Mechanisms of Action for Antimicrobial Peptides with Antibacterial and Antibiofilm Functions. Front. Microbiol. 2019, 10, 2866. [Google Scholar] [CrossRef] [Green Version]
- Assoni, L.; Milani, B.; Carvalho, M.R.; Nepomuceno, L.N.; Waz, N.T.; Guerra, M.E.S.; Converso, T.R.; Darrieux, M. Resistance Mechanisms to Antimicrobial Peptides in Gram-Positive Bacteria. Front. Microbiol. 2020, 11, 593215. [Google Scholar] [CrossRef]
- Strøm, M.B.; Haug, B.E.; Skar, M.L.; Stensen, W.; Stiberg, T.; Svendsen, J.S. The Pharmacophore of Short Cationic Antibacterial Peptides. J. Med. Chem. 2003, 46, 1567–1570. [Google Scholar] [CrossRef]
- Francolini, I.; Donelli, G. Prevention and control of biofilm-based medical-device-related infections. FEMS Immunol. Med. Microbiol. 2010, 59, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri, M.; Beyermann, M.; Dathe, M. Immobilization Reduces the Activity of Surface-Bound Cationic Antimicrobial Peptides with No Influence upon the Activity Spectrum. Antimicrob. Agents Chemother. 2009, 53, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- Cerruti, M.; Fissolo, S.; Carraro, C.; Ricciardi, C.; Majumdar, A.; Maboudian, R. Poly(ethylene glycol) Monolayer Formation and Stability on Gold and Silicon Nitride Substrates. Langmuir 2008, 24, 10646–10653. [Google Scholar] [CrossRef]
- Castro, V.; Rodríguez, H.; Albericio, F. CuAAC: An Efficient Click Chemistry Reaction on Solid Phase. ACS Comb. Sci. 2016, 18, 1–14. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Strøm, M.B.; Rekdal, Ø.; Svendsen, J.S. Antimicrobial activity of short arginine- and tryptophan-rich peptides. J. Pept. Sci. 2002, 8, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Haug, B.E.; Skar, M.L.; Svendsen, J.S. Bulky aromatic amino acids increase the antibacterial activity of 15-residue bovine lactoferricin derivatives. J. Pept. Sci. 2001, 7, 425–432. [Google Scholar] [CrossRef]
- Haug, B.E.; Svendsen, J.S. The role of tryptophan in the antibacterial activity of a 15-residue bovine lactoferricin peptide. J. Pept. Sci. 2001, 7, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Haug, B.E.; Stensen, W.; Stiberg, T.; Svendsen, J.S. Bulky Nonproteinogenic Amino Acids Permit the Design of Very Small and Effective Cationic Antibacterial Peptides. J. Med. Chem. 2004, 47, 4159–4162. [Google Scholar] [CrossRef] [PubMed]
- Haug, B.E.; Stensen, W.; Svendsen, J.S. Application of the Suzuki–Miyaura cross-coupling to increase antimicrobial potency generates promising novel antibacterials. Bioorg. Med. Chem. Lett. 2007, 17, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Förch, R.; Schönherr, H.; Jenkins, A.T.A. Surface Design: Applications in Bioscience and Nanotechnology; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Jones, C.W.; Morales, C.G.; Eltiste, S.L.; Yanchik-Slade, F.E.; Lee, N.R.; Nilsson, B.L. Capacity for increased surface area in the hydrophobic core of β -sheet peptide bilayer nanoribbons. J. Pept. Sci. 2021, 27, e3334. [Google Scholar] [CrossRef] [PubMed]
- Pauloehrl, T.; Welle, A.; Bruns, M.; Linkert, K.; Börner, H.G.; Bastmeyer, M.; Delaittre, G.; Barner-Kowollik, C. Spatially Controlled Surface Immobilization of Nonmodified Peptides. Angew. Chem. Int. Ed. 2013, 52, 9714–9718. [Google Scholar] [CrossRef] [PubMed]
- Bruenke, J.; Roschke, I.; Agarwal, S.; Riemann, T.; Greiner, A. Quantitative Comparison of the Antimicrobial Efficiency of Leaching versus Nonleaching Polymer Materials. Macromol. Biosci. 2016, 16, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dathe, M.; Nikolenko, H.; Klose, J.; Bienert, M. Cyclization Increases the Antimicrobial Activity and Selectivity of Arginine- and Tryptophan-Containing Hexapeptides. Biochemistry 2004, 43, 9140–9150. [Google Scholar] [CrossRef]
- Herzberg, M.; Berglin, M.; Eliahu, S.; Bodin, L.; Agrenius, K.; Zlotkin, A.; Svenson, J. Efficient Prevention of Marine Biofilm Formation Employing a Surface-Grafted Repellent Marine Peptide. ACS Appl. Bio Mater. 2021, 4, 3360–3373. [Google Scholar] [CrossRef]
- Flemming, K.; Klingenberg, C.; Cavanagh, P.; Sletteng, M.; Stensen, W.; Svendsen, J.S.; Flaegstad, T.; Flægstad, T. High in vitro antimicrobial activity of synthetic antimicrobial peptidomimetics against staphylococcal biofilms. J. Antimicrob. Chemother. 2008, 63, 136–145. [Google Scholar] [CrossRef]
- European Committee for Antimicrobial Susceptibility Testing (EUCAST) of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID). Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin. Microbiol. Infect. 2003, 9, ix–xv.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Activity (MIC in µg/mL) | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Sequence | Net Charge | Mw | S. aureus | S. epidermidis | E. coli | P. aeruginosa |
| 1a | N3-PEG200-RWWRF | 2+ | 1188.49 | 64 | 32 | 256 | 256 |
| 1b | N3-PEG400-RWWRF | 2+ | 1388.49 | 128 | 128 | >256 | >256 |
| 1c | RWWRFK(N3) | 3+ | 1003.19 | 32 | 16 | 64 | 64 |
| 1d | c(RWWRFK(N3)) | 2+ | 986.16 | 8 | 8 | 64 | 128 |
| 2a | N3-PEG200-RBBRF | 2+ | 1262.53 | 8 | 8 | 64 | 64 |
| 2b | N3-PEG400-RBBRF | 2+ | 1462.53 | 32 | 16 | 128 | 256 |
| 2c | RBBRFK(N3) | 3+ | 1077.31 | 8 | 4 | 8 | 16 |
| 2d | c(RBBRFK(N3)) | 2+ | 1060.28 | 4 | 2 | 64 | 256 |
| 1a | 1b | 1c | 1d | 2a | 2b | 2c | 2d | Control |
|---|---|---|---|---|---|---|---|---|
| 49.1 (2.7) | 52.2 (2.5) | 50.0 (3.2) | 49.6 (2.1) | 54.7 (2.4) | 53.5 (2.3) | 54.9 (3.4) | 54.0 (2.5) | 39.6 (2.8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karlsen, E.A.; Stensen, W.; Juskewitz, E.; Svenson, J.; Berglin, M.; Svendsen, J.S.M. Anti-Colonization Effect of Au Surfaces with Self-Assembled Molecular Monolayers Functionalized with Antimicrobial Peptides on S. epidermidis. Antibiotics 2021, 10, 1516. https://doi.org/10.3390/antibiotics10121516
Karlsen EA, Stensen W, Juskewitz E, Svenson J, Berglin M, Svendsen JSM. Anti-Colonization Effect of Au Surfaces with Self-Assembled Molecular Monolayers Functionalized with Antimicrobial Peptides on S. epidermidis. Antibiotics. 2021; 10(12):1516. https://doi.org/10.3390/antibiotics10121516
Chicago/Turabian StyleKarlsen, Eskil André, Wenche Stensen, Eric Juskewitz, Johan Svenson, Mattias Berglin, and John Sigurd Mjøen Svendsen. 2021. "Anti-Colonization Effect of Au Surfaces with Self-Assembled Molecular Monolayers Functionalized with Antimicrobial Peptides on S. epidermidis" Antibiotics 10, no. 12: 1516. https://doi.org/10.3390/antibiotics10121516
APA StyleKarlsen, E. A., Stensen, W., Juskewitz, E., Svenson, J., Berglin, M., & Svendsen, J. S. M. (2021). Anti-Colonization Effect of Au Surfaces with Self-Assembled Molecular Monolayers Functionalized with Antimicrobial Peptides on S. epidermidis. Antibiotics, 10(12), 1516. https://doi.org/10.3390/antibiotics10121516