
Genomic Investigation of Methicillin-Resistant Staphylococcus aureus ST113 Strains Isolated from Tertiary Care Hospitals in Pakistan

 ,
,  , ,
, , 

Abstract
:1. Introduction
2. Results
2.1. Phenotypic Antibiotic Resistance of the Isolates
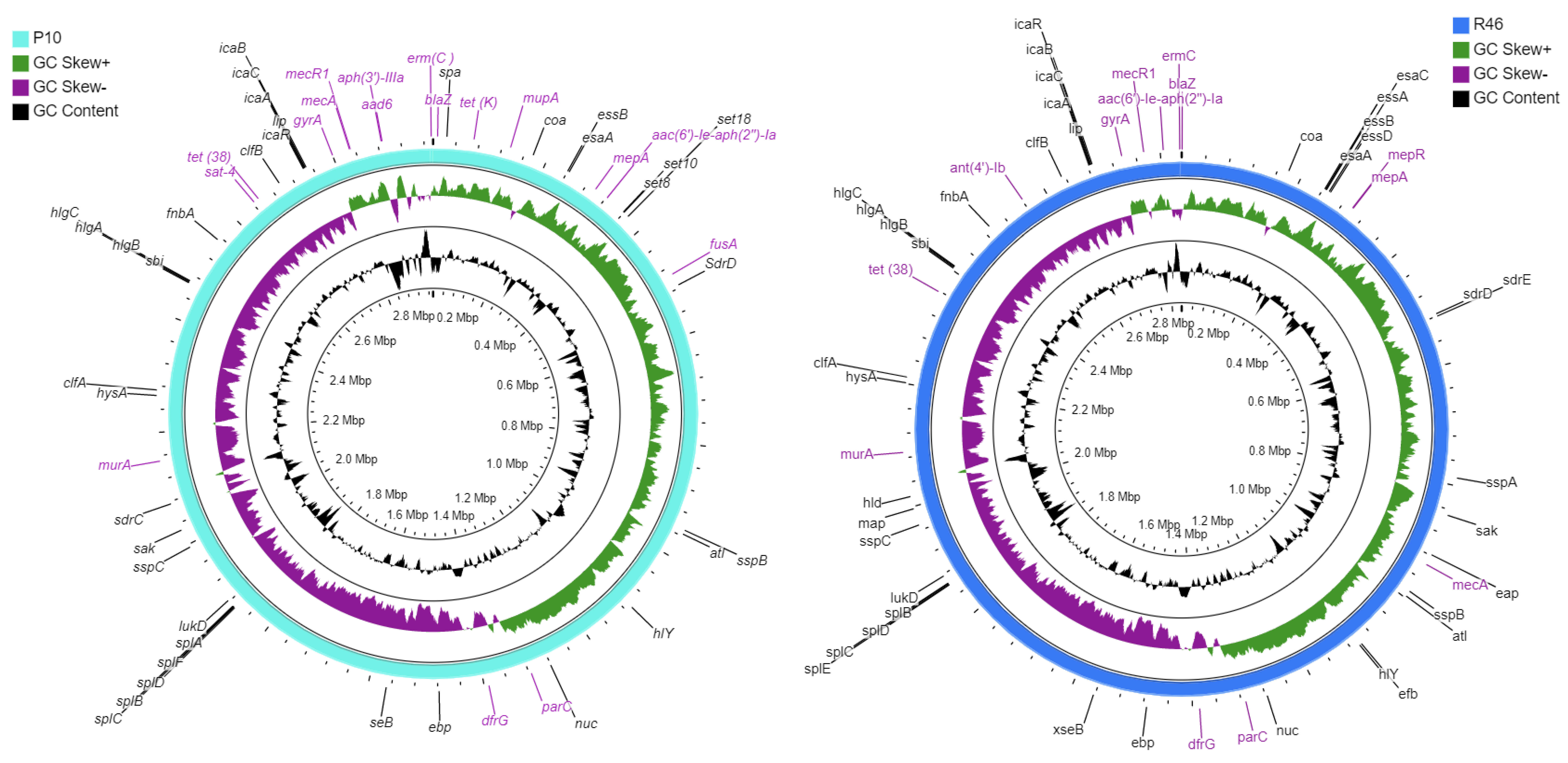
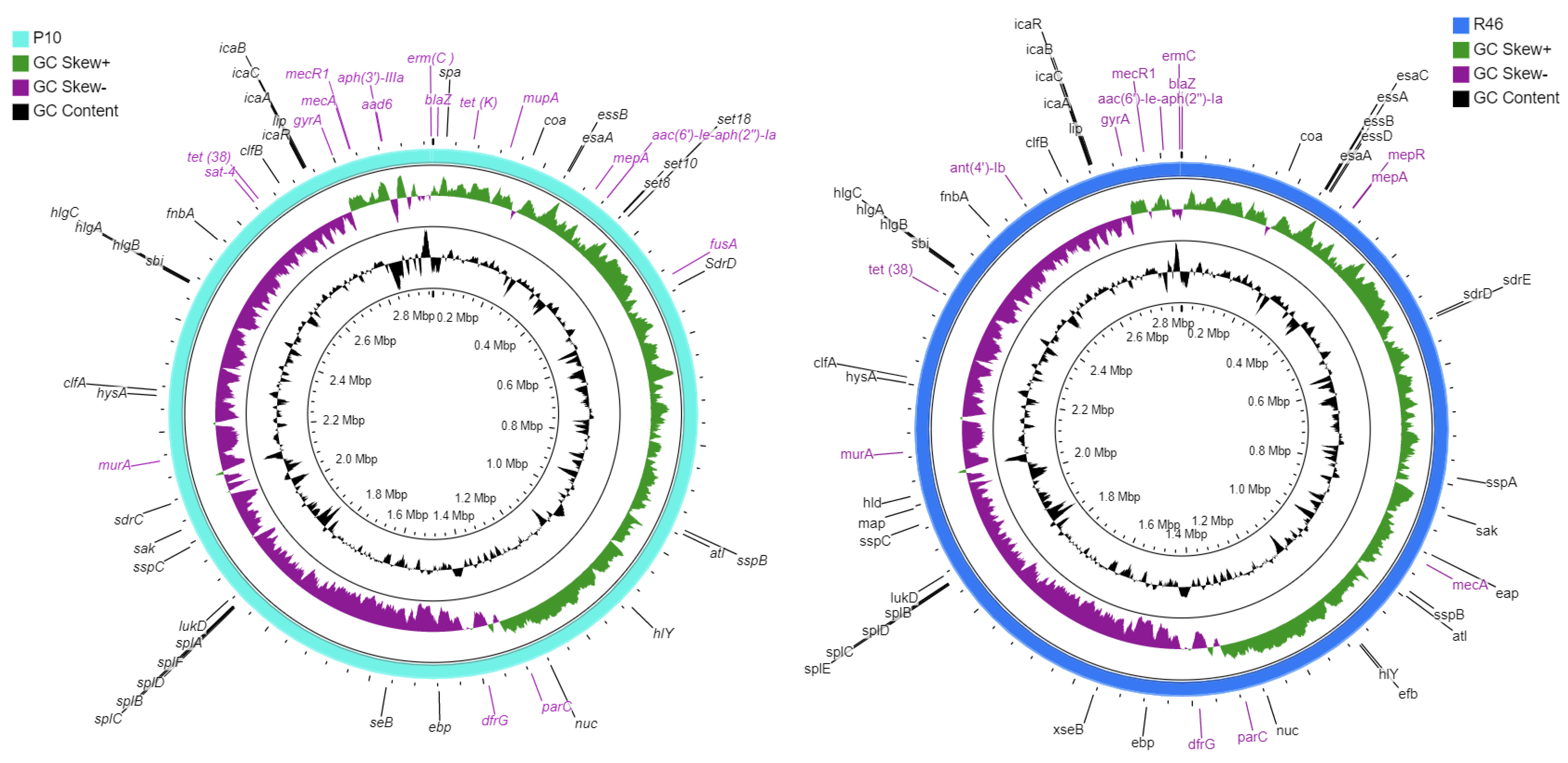
2.2. Genomic Characteristics
2.3. Genome-Based Typing and Mobile Genetic Elements
2.4. Genes-Associated with Antibiotic Resistance and Virulence
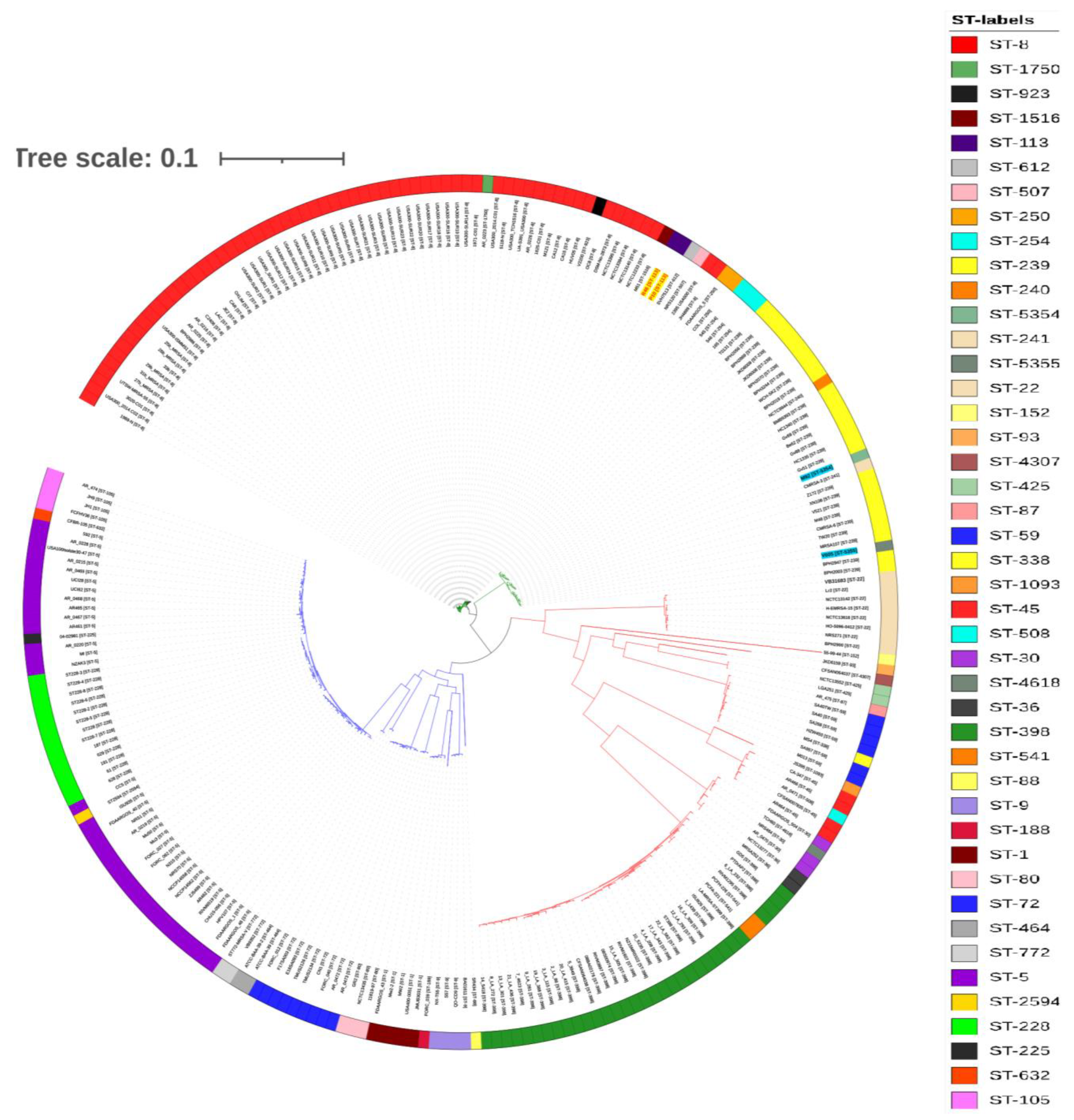
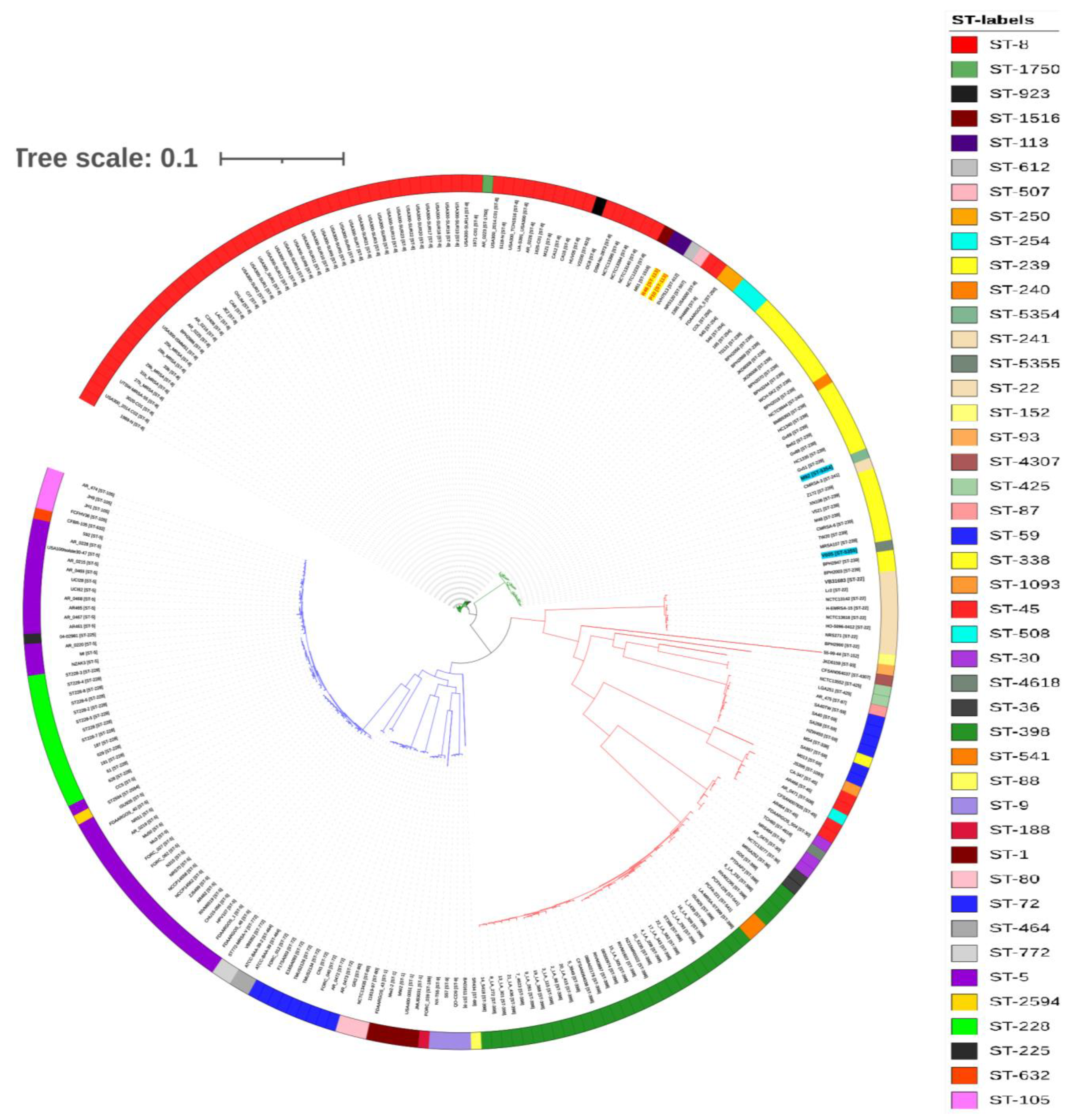
2.5. Phylogenetic Analysis and Comparison of Antibiotic-Resistance Determinants
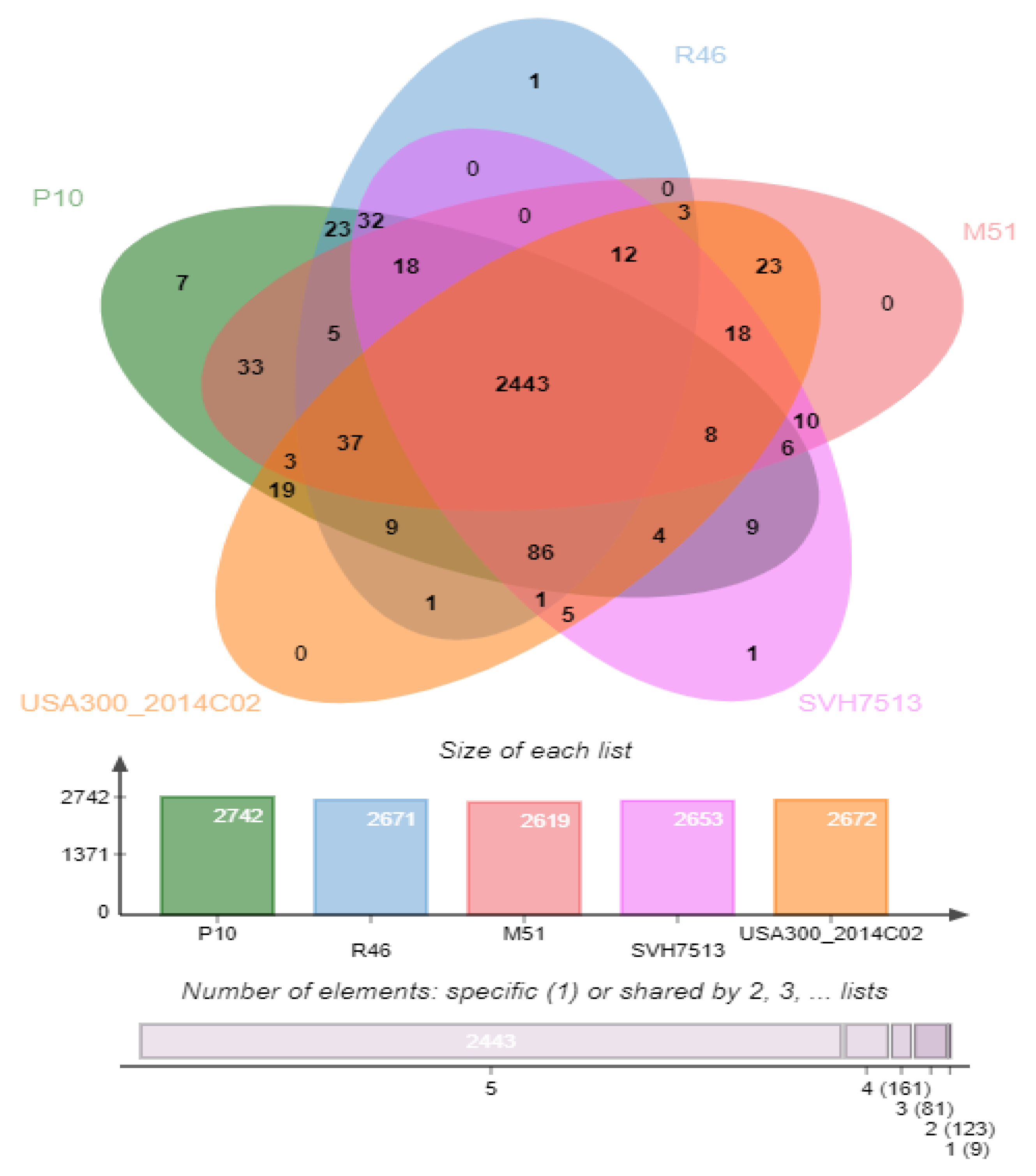
2.6. Genome-Wide Comparison of Clusters of Orthologous Groups (COGs)
3. Discussion
4. Conclusions
5. Methods
5.1. Bacterial Isolates Collection and Antibiotic Susceptibility Testing
5.2. Whole-Genome Sequence Analysis and Molecular Typing
5.3. Identification of Plasmids, Prophages, and Genes Associated with Antibiotic Resistance and Virulence
5.4. MLST and SNP Based Phylogenetic Analysis
5.5. Comparative Analysis of Whole-Genome Orthologous Clusters
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Liang, Y.; Tu, C.; Tan, C.; El-Sayed Ahmed, M.A.E.-G.; Dai, M.; Xia, Y.; Liu, Y.; Zhong, L.-L.; Shen, C.; Chen, G.; et al. Antimicrobial resistance, virulence genes profiling and molecular relatedness of methicillin-resistant Staphylococcus aureus strains isolated from hospitalized patients in Guangdong Province, China. Infect. Drug Resist. 2019, 12, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Rong, D.; Wu, Q.; Xu, M.; Zhang, J.; Yu, S. Prevalence, Virulence Genes, Antimicrobial Susceptibility, and Genetic Diversity of Staphylococcus aureus from Retail Aquatic Products in China. Front. Microbiol. 2017, 8, 714. [Google Scholar] [CrossRef] [Green Version]
- Ullah, N.; Raza, T.; Dar, H.A.; Shehroz, M.; Zaheer, T.; Naz, K.; Ali, A. Whole-genome sequencing of a new sequence type (ST5352) strain of community-acquired methicillin-resistant Staphylococcus aureus from a hospital in Pakistan. J. Glob. Antimicrob. Resist. 2019, 19, 161–163. [Google Scholar] [CrossRef] [PubMed]
- Brown-Jaque, M.; Rodriguez Oyarzun, L.; Cornejo-Sánchez, T.; Martín-Gomez, M.T.; Gartner, S.; De Gracia, J.; Rovira, S.; Alvarez, A.; Jofre, J.; González-López, J.J.; et al. Detection of Bacteriophage Particles Containing Antibiotic Resistance Genes in the Sputum of Cystic Fibrosis Patients. Front. Microbiol. 2018, 9, 856. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Amirsoleimani, A.; Brion, G.M.; Diene, S.M.; Francois, P.; Richard, E.M. Prevalence and characterization of Staphylococcus aureus in wastewater treatment plants by whole genomic sequencing. Water Res. 2019, 158, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003, 48, 1429–1449. [Google Scholar] [CrossRef]
- Kong, C.; Neoh, H.-M.; Nathan, S. Targeting Staphylococcus aureus Toxins: A Potential form of Anti-Virulence Therapy. Toxins 2016, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Watkins, R.R.; David, M.; Salata, R.A. Current concepts on the virulence mechanisms of meticillin-resistant Staphylococcus aureus. J. Med. Microbiol. 2012, 61, 1179–1193. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, D.C.; Tomasz, A.; de Lencastre, H. Secrets of success of a human pathogen: Molecular evolution of pandemic clones of meticillin-resistant Staphylococcus aureus. Lancet Infect. Dis. 2002, 2, 180–189. [Google Scholar] [CrossRef]
- Xu, Z.; Misra, R.; Jamrozy, D.; Paterson, G.; Cutler, R.R.; Holmes, M.A.; Gharbia, S.; Mkrtchyan, H.V. Whole Genome Sequence and Comparative Genomics Analysis of Multi-drug Resistant Environmental Staphylococcus epidermidis ST59. G3 Genes Genomes Genet. 2018, 8, 2225–2230. [Google Scholar] [CrossRef] [Green Version]
- Baig, S.; Johannesen, T.B.; Overballe-Petersen, S.; Larsen, J.; Larsen, A.R.; Stegger, M. Novel SCCmec type XIII (9A) identified in an ST152 methicillin-resistant Staphylococcus aureus. Infect. Genet. Evol. 2018, 61, 74–76. [Google Scholar] [CrossRef]
- Lim, K.T.; Yeo, C.C.; Suhaili, Z.; Thong, K.L. Comparison of Methicillin-Resistant and Methicillin-Sensitive Staphylococcus aureus Strains Isolated from a Tertiary Hospital in Terengganu, Malaysia. Jpn. J. Infect. Dis. 2012, 65, 502–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Li, F.; Liu, D.; Xue, H.; Zhao, X. Novel type XII staphylococcal cassette chromosome mec harboring a new cassette chromosome recombinase, CcrC2. Antimicrob. Agents Chemother. 2015, 59, 7597–7601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, G.I.; Shore, A.; Corcoran, S.; Tecklenborg, S.; Coleman, D.; O’Connell, B. Emergence of Hospital- and Community-Associated Panton-Valentine Leukocidin-Positive Methicillin-Resistant Staphylococcus aureus Genotype ST772-MRSA-V in Ireland and Detailed Investigation of an ST772-MRSA-V Cluster in a Neonatal Intensive Care Unit. J. Clin. Microbiol. 2011, 50, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Marasa, B.; Sung, K.; Nawaz, M. Genotypic Characterization of Clinical Isolates of Staphylococcus aureus from Pakistan. Pathogens 2021, 10, 918. [Google Scholar] [CrossRef]
- Shabir, S.; Hardy, K.J.; Abbasi, W.S.; McMurray, C.L.; Malik, S.A.; Wattal, C.; Hawkey, P.M. Epidemiological typing of meticillin-resistant Staphylococcus aureus isolates from Pakistan and India. J. Med. Microbiol. 2010, 59, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Zafar, A.; Stone, M.; Ibrahim, S.; Parveen, Z.; Hasan, Z.; Khan, E.; Hasan, R.; Wain, J.; Bamford, K. Prevalent genotypes of meticillin-resistant Staphylococcus aureus: Report from Pakistan. J. Med. Microbiol. 2011, 60, 56–62. [Google Scholar] [CrossRef]
- Madzgalla, S.; Syed, M.A.; Khan, M.A.; Rehman, S.S.; Müller, E.; Reissig, A.; Ehricht, R.; Monecke, S. Molecular characterization of Staphylococcus aureus isolates causing skin and soft tissue infections in patients from Malakand, Pakistan. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 35, 1541–1547. [Google Scholar] [CrossRef]
- Arfat, Y. Genotyping of Methicillin Resistant Staphylococcus aureus (MRSA) from Local Hospital of Rawalpindi/Islamabad, Pakistan; Quaid-i-Azam University Islamabad: Islamabad, Pakistan, 2013. [Google Scholar]
- Nguyen, D.B.; Lessa, F.C.; Belflower, R.; Mu, Y.; Wise, M.; Nadle, J.; Bamberg, W.M.; Petit, S.; Ray, S.M.; Harrison, L.H.; et al. Invasive methicillin-resistant Staphylococcus aureus infections among patients on chronic dialysis in the United States, 2005–2011. Clin. Infect. Dis. 2013, 57, 1393–1400. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, N.A.; Hussein, N.R. Staphylococcus aureus: An Overview of Discovery, Characteristics, Epidemiology, Virulence Factors and Antimicrobial Sensitivity. Eur. J. Mol. Clin. Med. 2021, 8, 1160–1183. [Google Scholar]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preeja, P.; Kumar, S.; Shetty, V. Prevalence and Characterization of Methicillin-Resistant Staphylococcus aureus from Community- and Hospital-Associated Infections: A Tertiary Care Center Study. Antibiotics 2021, 10, 197. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Baba, T.; Hiramatsu, K.; Schneewind, O. Prophages of Staphylococcus aureus Newman and their contribution to virulence. Mol. Microbiol. 2006, 62, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Kashif, A.; McClure, J.-A.; Lakhundi, S.; Pham, M.; Chen, S.; Conly, J.M.; Zhang, K. Staphylococcus aureus ST398 Virulence Is Associated With Factors Carried on Prophage ϕSa3. Front. Microbiol. 2019, 10, 2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.; Costa, S.; Serrano, M.; Oliveira, K.; Trigueiro, G.; Pomba, C.; Couto, I. Clonal Lineages, Antimicrobial Resistance, and PVL Carriage of Staphylococcus aureus Associated to Skin and Soft-Tissue Infections from Ambulatory Patients in Portugal. Antibiotics 2021, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Stevens, D.L.; Hamilton, S.M.; Parimon, T.; Ma, Y.; Kearns, A.M.; Ellis, R.W.; Bryant, A.E.; Fatal, S. aureus Hemorrhagic Pneumonia: Genetic Analysis of a Unique Clinical Isolate Producing both PVL and TSST-1. PLoS ONE 2011, 6, e27246. [Google Scholar] [CrossRef]
- Asghar, A.H. Molecular characterization of methicillin-resistant Staphylococcus aureus isolated from tertiary care hospitals. Pak. J. Med. Sci. 2014, 30, 698. [Google Scholar] [CrossRef]
- Campbell, S.J.; Deshmukh, H.S.; Nelson, C.L.; Bae, I.-G.; Stryjewski, M.; Federspiel, J.J.; Tonthat, G.T.; Rude, T.H.; Barriere, S.L.; Corey, R.; et al. Genotypic Characteristics of Staphylococcus aureus Isolates from a Multinational Trial of Complicated Skin and Skin Structure Infections. J. Clin. Microbiol. 2008, 46, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.A.; Ali, A.; Tharmalingam, N.; Mylonakis, E.; Zahra, R. First report of mecC gene in clinical methicillin resistant S. aureus (MRSA) from tertiary care hospital Islamabad, Pakistan. J. Infect. Public Health 2020, 13, 1501–1507. [Google Scholar] [CrossRef]
- Mongodin, E.; Bajolet, O.; Cutrona, J.; Bonnet, N.; Dupuit, F.; Puchelle, E.; de Bentzmann, S. Fibronectin-Binding Proteins of Staphylococcus aureus Are Involved in Adherence to Human Airway Epithelium. Infect. Immun. 2002, 70, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planet, P.J. Life after USA300: The rise and fall of a superbug. J. Infect. Dis. 2017, 215 (Suppl. 1), S71–S77. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, A.; Dua, P.; Ghosh, C. Biochemical and Molecular Analysis of Staphylococcus aureus Clinical Isolates from Hospitalized Patients. Can. J. Infect. Dis. Med. Microbiol. 2016, 2016, 9041636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pournajaf, A.; Ardebili, A.; Goudarzi, L.; Khodabandeh, M.; Narimani, T.; Abbaszadeh, H. PCR-based identification of methicillin–resistant Staphylococcus aureus strains and their antibiotic resistance profiles. Asian Pac. J. Trop. Biomed. 2014, 4, S293–S297. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics 2009, 25, 2071–2073. [Google Scholar] [CrossRef]
- Grant, J.R.; Arantes, A.S.; Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 2012, 13, 202. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- McArthur, A.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The Comprehensive Antibiotic Resistance Database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.C.F.; Ahrenfeldt, J.; Cisneros, J.L.B.; Jurtz, V.I.; Larsen, M.V.; Hasman, H.; Aarestrup, F.; Lund, O. A Bacterial Analysis Platform: An Integrated System for Analysing Bacterial Whole Genome Sequencing Data for Clinical Diagnostics and Surveillance. PLoS ONE 2016, 11, e0157718. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.; Lund, O. Solving the Problem of Comparing Whole Bacterial Genomes across Different Sequencing Platforms. PLoS ONE 2014, 9, e104984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | P10 | R46 | ||
|---|---|---|---|---|
| PR | GR | PR | GR | |
| Ampicillin | R | blaZ, mecA, mecR1 | R | blaZ, mecA, mecR1 |
| Methicillin | R | R | ||
| Oxacillin | R | R | ||
| Gentamicin | R | aac(6′)-Ie-aph(2″)-Ia, aph(3’)-IIIa,aad(6) | R | aac(6′)-Ie-aph(2″)-Ia, ant(4’)-Ib |
| Streptomycin | R | R | ||
| Erythromycin | R | erm(C) | R | erm(C) |
| Clindamycin | R | R | ||
| Linezolid | R | R | ||
| Cefixime | R | ND | R | ND |
| Cefepime | R | ND | S | ND |
| Meropenem | R | ND | R | ND |
| Chloramphenicol | S | ND | S | ND |
| Tetracycline | R | mepA, tet(38), tet(K), mepR | R | mepA, tet(38), mepR |
| Ciprofloxacin | R | gyrA, parC | R | gyrA, parC |
| Rifampicin | S | ND | R | ND |
| Fusidic acid | R | fusA | R | ND |
| Vancomycin | S | ND | S | ND |
| Trimethoprim | ND | dfrG | ND | dfrG |
| Streptothrin | ND | sat-4 | ND | ND |
| Mupirocin | ND | mupA | ND | ND |
| Fosfomycin | ND | murA | ND | murA |
| Characteristics | P10 | R46 |
|---|---|---|
| Genome size (bp) | 2,955,291 | 2,822,631 |
| Contigs | 90 | 72 |
| GC content % | 32.7 | 32.7 |
| N50 | 84,395 | 84,399 |
| N75 | 52,815 | 50,022 |
| L50 | 10 | 11 |
| Largest Contig (bp) | 294,580 | 261,096 |
| N50 | 84,395 | 84,399 |
| No. of CDS | 3031 | 2827 |
| No. of tRNA | 54 | 55 |
| No. of rRNA | 9 | 10 |
| ST | 113 | 113 |
| SCCmec type | IV | IV |
| spa-type | t064 | unknown |
| NCBI Accession number | JAHHEA000000000.1 | JAHKSM000000000.1 |
| Strain | Plasmid | Total Length | Most Similar Plasmid | % Similarity | Accession Number |
|---|---|---|---|---|---|
| P10 | 1 | 2375 bp | pI5S5 | 99.78 | HE579068.1 |
| 2 | 2855 bp | unnamed1 | 99.72 | CP030577.1 | |
| R46 | 1 | 2494 bp | Staphylococcus epidermidis isolate BPH0662 plasmid: 1 | 99.83 | LT614820.1 |
| 2 | 2250 bp | pRM27 | 100 | KT780704.1 |
| Strain | Region | Region Length | Total Proteins | Phage Hit Proteins | GC % | Specific Keywords | Most Common Phage |
|---|---|---|---|---|---|---|---|
| P10 | 1 | 48.4 kbp | 64 | 64 | 35.02 | recombinase, terminase, portal, head, capsid, tail | Staphy_SA13_NC_021863 |
| 2 | 74.2 kbp | 79 | 69 | 32.84 | tail, capsid, head, portal, integrase | Staphy_phi2958PVL_NC_011344 | |
| 3 | 42 kbp | 46 | 29 | 32.66 | integrase, portal, protease, capsid, head, tail | Staphy_phiN315_NC_004740 | |
| R46 | 1 | 55.6 kb | 69 | 67 | 32.79 | integrase, terminase, portal, protease, head, tail | Staphy_phiNM3_NC_008617 |
| 2 | 71.4 kb | 76 | 68 | 32.77 | tail, capsid, head, portal, integrase | Staphy_phi2958PVL_NC_011344 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ullah, N.; Dar, H.A.; Naz, K.; Andleeb, S.; Rahman, A.; Saeed, M.T.; Hanan, F.; Bae, T.; Ali, A. Genomic Investigation of Methicillin-Resistant Staphylococcus aureus ST113 Strains Isolated from Tertiary Care Hospitals in Pakistan. Antibiotics 2021, 10, 1121. https://doi.org/10.3390/antibiotics10091121
Ullah N, Dar HA, Naz K, Andleeb S, Rahman A, Saeed MT, Hanan F, Bae T, Ali A. Genomic Investigation of Methicillin-Resistant Staphylococcus aureus ST113 Strains Isolated from Tertiary Care Hospitals in Pakistan. Antibiotics. 2021; 10(9):1121. https://doi.org/10.3390/antibiotics10091121
Chicago/Turabian StyleUllah, Nimat, Hamza Arshad Dar, Kanwal Naz, Saadia Andleeb, Abdur Rahman, Muhammad Tariq Saeed, Fazal Hanan, Taeok Bae, and Amjad Ali. 2021. "Genomic Investigation of Methicillin-Resistant Staphylococcus aureus ST113 Strains Isolated from Tertiary Care Hospitals in Pakistan" Antibiotics 10, no. 9: 1121. https://doi.org/10.3390/antibiotics10091121
APA StyleUllah, N., Dar, H. A., Naz, K., Andleeb, S., Rahman, A., Saeed, M. T., Hanan, F., Bae, T., & Ali, A. (2021). Genomic Investigation of Methicillin-Resistant Staphylococcus aureus ST113 Strains Isolated from Tertiary Care Hospitals in Pakistan. Antibiotics, 10(9), 1121. https://doi.org/10.3390/antibiotics10091121