

Embedded-AMP: A Multi-Thread Computational Method for the Systematic Identification of Antimicrobial Peptides Embedded in Proteome Sequences

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
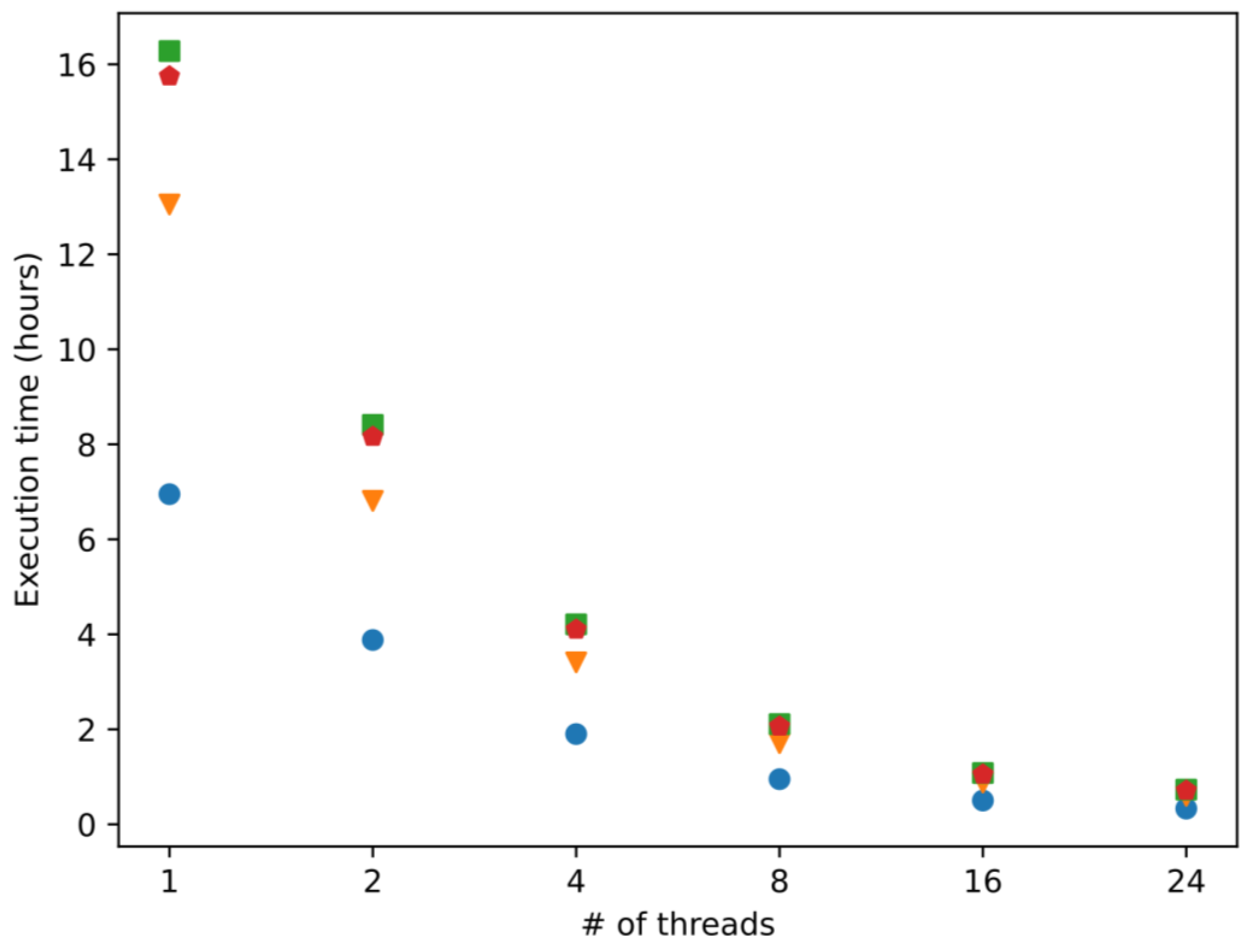
2.1. Computation Efficiency
2.2. Scalability and Computational Efficiency
2.3. Peptide Sequence Frequency
2.4. Testing Predictions of Embedded AMP on Shrimp Proteome
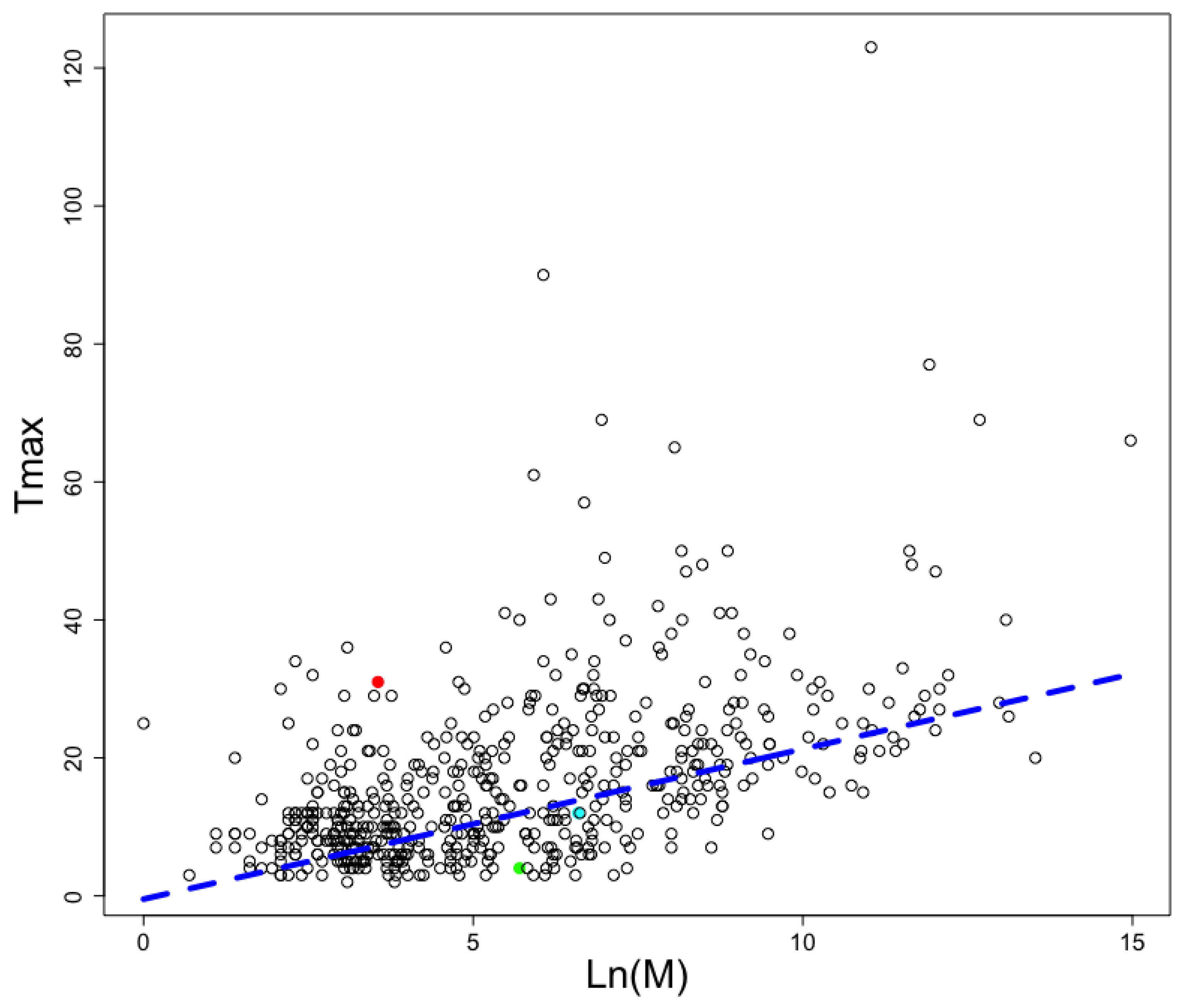
2.5. Predicting AMP in Different Animals with Different Longevities
2.6. Evaluating Predictions of Embedded AMPs with Known Cases
3. Discussion
4. Materials and Methods
4.1. Equipment and Libraries Used
4.2. Estimating the Problem Size
4.3. K-Mer Detection and Counting
| Algorithm 1:K-mers extractor |
| Input: Proteome, kmin, //k-mers minimun length kmax //k-mers maximum length Output: unique k-mers Begin for i = kmin to kmax do add(k-mer, proteome) sort(k-mers) Delete Duplicates End |
| Algorithm 2: Delete Duplicates |
| Input: k-mers to compare BEGIN total <- length(k-mers) for j = 0, k = 1 to total-1 do if k-mer[j] ≠ k-mer[k] then j = j + 1 k-mer[j] = k-mer[k] No_of_different_k-mers = j End |
4.4. Prediction of AMP
4.5. Pruning of k-Mers
| Algorithm 3: Pruning |
| Input: k-mers derived from a specific sequence in the proteome Output: groups//groups of continuous k-mers within the input sequence BEGIN Sort k-mers by their initial position in the given proteome sequence total <- LENGTH(k-mers)//total number of k-mers create the empty set groups whose elements are k-group//groups contain k-group, //every fragment of the proteome covered by consecutive k-mers predicted as AMPs. j <- 1 k-group.first <- k-mer[0].first //the initial position of the left most k-mer while j < total do k-group.last <- k-mer[j − 1].last//the last position of the j − 1 k-mer if k-mer[j] NOT Embedded(k-group) if k-mer[j] Connected(k-group) OR k-mer[j] Intersects(k-group) then k-last <- k-mer[j] else Add k-group to groups k-group.first <- k-mer[j].first k-group.last <- k-mer[j].last END |
| Algorithm 4: Embedded |
| Input: k-mer[i], k-mer[j] Output: found //k-mer i is embedded within k-mer j BEGIN found <- FALSE if k-mer[i].first ≥ k-mer[j].first AND k-mer[i].last ≤ k-mer[j].last found <- TRUE END |
| Algorithm 5: Connected |
| Input: k-mer[i], k-mer[j] Output: connected BEGIN connected <- FALSE if k-mer[i].first < k-mer[j].first AND k-mer[i].last == k-mer[j].last OR k-mer[i].first == (k-mer[j].last + 1) then connected <- TRUE END |
| Algorithm 6: Intersects |
| Input: k-mer[i], k-mer[j] Output: intersects BEGIN intersects <- FALSE if k-mer[i].first <= k-mer[j].first AND k-mer[i].last < k-mer[j].last then intersects <- TRUE END |
4.6. Tool Availability
4.7. Proteome Sequences
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bahar, A.A.; Ren, D. Antimicrobial Peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiro, K.G.N.; Rodrigues, G.; Monges, B.E.D.; Cardoso, M.H.; Franco, O.L. Bioactive Peptides Against Fungal Biofilms. Front. Microbiol. 2019, 10, 2169. [Google Scholar] [CrossRef]
- Pen, G.; Yang, N.; Teng, D.; Mao, R.; Hao, Y.; Wang, J. A Review on the Use of Antimicrobial Peptides to Combat Porcine Viruses. Antibiotics 2020, 9, 801. [Google Scholar] [CrossRef]
- Vale, N.; Aguiar, L.; Egomes, P. Antimicrobial peptides: A new class of antimalarial drugs? Front. Pharmacol. 2014, 5, 275. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Singh, S.; Nagaraj, R.; Vaidya, T. Induction of autophagic cell death in Leishmania donovani by antimicrobial peptides. Mol. Biochem. Parasitol. 2003, 127, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyotl, E.A.P.; Palacios, J.B.; Muciño, G.; Moreno-Blas, D.; Costas, M.; Montes, T.M.; Diener, C.; Uribe-Carvajal, S.; Massieu, L.; Castro-Obregón, S.; et al. Antimicrobial Peptide against Mycobacterium Tuberculosis That Activates Autophagy Is an Effective Treatment for Tuberculosis. Pharmaceutics 2020, 12, 1071. [Google Scholar] [CrossRef] [PubMed]
- Rekha, R.S.; Muvva, S.J.R.; Wan, M.; Raqib, R.; Bergman, P.; Brighenti, S.; Gudmundsson, G.H.; Agerberth, B. Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy 2015, 11, 1688–1699. [Google Scholar] [CrossRef] [Green Version]
- Branco, P.; Francisco, D.; Chambon, C.; Hébraud, M.; Arneborg, N.; Almeida, M.G.; Caldeira, J.; Albergaria, H. Identification of novel GAPDH-derived antimicrobial peptides secreted by Saccharomyces cerevisiae and involved in wine microbial interactions. Appl. Microbiol. Biotechnol. 2013, 98, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Ponpuak, M.; Davis, A.S.; Roberts, E.A.; Delgado, M.A.; Dinkins, C.; Zhao, Z.; Virgin, H.W.; Kyei, G.B.; Johansen, T.; Vergne, I.; et al. Delivery of Cytosolic Components by Autophagic Adaptor Protein p62 Endows Autophagosomes with Unique Antimicrobial Properties. Immunity 2010, 32, 329–341. [Google Scholar] [CrossRef]
- Aguilera-Mendoza, L.; Marrero-Ponce, Y.; Beltran, J.A.; Ibarra, R.T.; Guillen-Ramirez, H.; Brizuela, C.A. Graph-based data integration from bioactive peptide databases of pharmaceutical interest: Toward an organized collection enabling visual network analysis. Bioinformatics 2019, 35, 4739–4747. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.C.R.; Maasch, J.R.M.A.; de la Fuente-Nunez, C. Accelerating antibiotic discovery through artificial intelligence. Commun. Biol. 2021, 4, 1050. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, M.H.; Orozco, R.M.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.D.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Fan, L.; Sun, J.; Lao, X.; Zheng, H. Computational resources and tools for antimicrobial peptides. J. Pept. Sci. 2016, 23, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Beltran, J.A.; Del Rio, G.; Brizuela, C.A. An automatic representation of peptides for effective antimicrobial activity classification. Comput. Struct. Biotechnol. J. 2020, 18, 455–463. [Google Scholar] [CrossRef]
- García-Jacas, C.R.; Pinacho-Castellanos, S.A.; García-González, L.A.; A Brizuela, C. Do deep learning models make a difference in the identification of antimicrobial peptides? Brief. Bioinform. 2022, 23, bbac094. [Google Scholar] [CrossRef]
- Fingerhut, L.C.H.W.; Miller, D.J.; Strugnell, J.M.; Daly, N.L.; Cooke, I.R. ampir: An R package for fast genome-wide prediction of antimicrobial peptides. Bioinformatics 2020, 36, 5262–5263. [Google Scholar] [CrossRef]
- Santos-Júnior, C.D.; Pan, S.; Zhao, X.-M.; Coelho, L.P. Macrel: Antimicrobial peptide screening in genomes and metagenomes. Peerj 2020, 8, e10555. [Google Scholar] [CrossRef]
- Yoo, W.G.; Lee, J.H.; Shin, Y.; Shim, J.-Y.; Jung, M.; Kang, B.-C.; Oh, J.; Seong, J.; Lee, H.K.; Kong, H.S.; et al. Antimicrobial peptides in the centipede Scolopendra subspinipes mutilans. Funct. Integr. Genom. 2014, 14, 275–283. [Google Scholar] [CrossRef]
- Kim, I.-W.; Lee, J.H.; Subramaniyam, S.; Yun, E.-Y.; Kim, I.; Park, J.; Hwang, J.S. De Novo Transcriptome Analysis and Detection of Antimicrobial Peptides of the American Cockroach Periplaneta americana (Linnaeus). PLoS ONE 2016, 11, e0155304. [Google Scholar] [CrossRef]
- Robertson, L.S.; Cornman, R.S. Transcriptome resources for the frogs Lithobates clamitans and Pseudacris regilla, emphasizing antimicrobial peptides and conserved loci for phylogenetics. Mol. Ecol. Resour. 2013, 14, 178–183. [Google Scholar] [CrossRef]
- Wei, D.; Tian, C.-B.; Liu, S.-H.; Wang, T.; Smagghe, G.; Jia, F.-X.; Dou, W.; Wang, J.-J. Transcriptome analysis to identify genes for peptides and proteins involved in immunity and reproduction from male accessory glands and ejaculatory duct of Bactrocera dorsalis. Peptides 2016, 80, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A Library for Support Vector Machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Intel. Compile Cross-Architecture: Intel® oneAPI DPC++/C++ Compiler. Available online: https://www.intel.com/content/www/us/en/developer/tools/oneapi/dpc-compiler.html#gs.mn7agb (accessed on 4 January 2023).
- GCC. GCC, the GNU Compiler Collection—GNU Project. Available online: https://gcc.gnu.org/ (accessed on 4 January 2023).
- del Rio, G.; Perez, M.A.T.; Brizuela, C.A. Antimicrobial peptides with cell-penetrating activity as prophylactic and treatment drugs. Biosci. Rep. 2022, 42, BSR20221789. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Yoshimori, T. Autophagy and Longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [CrossRef]
- Tacutu, R.; Thornton, D.; Johnson, E.; Budovsky, A.; Barardo, D.; Craig, T.; Diana, E.; Lehmann, G.; Toren, D.; Wang, J.; et al. Human Ageing Genomic Resources: New and updated databases. Nucleic Acids Res. 2017, 46, D1083–D1090. [Google Scholar] [CrossRef]
- Santos, M.F.D.S.; Freitas, C.S.; da Costa, G.C.V.; Pereira, P.R.; Paschoalin, V.M.F. Identification of Antibacterial Peptide Candidates Encrypted in Stress-Related and Metabolic Saccharomyces cerevisiae Proteins. Pharmaceuticals 2022, 15, 163. [Google Scholar] [CrossRef]
- He, H.; Ma, Y. Imbalanced Learning: Foundations, Algorithms, and Applications; Wiley-IEEE Press: Hoboken, NJ, USA, 2013. [Google Scholar]
- Leevy, J.; Khoshgoftaar, T.M.; Bauder, R.A.; Seliya, N. A survey on addressing high-class imbalance in big data. J. Big Data 2018, 5, 42. [Google Scholar] [CrossRef]
- Alonso, S.; Pethe, K.; Russell, D.G.; Purdy, G.E. Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. USA 2007, 104, 6031–6036. [Google Scholar] [CrossRef]
- Albergaria, H.; Francisco, D.; Gori, K.; Arneborg, N.; Gírio, F. Saccharomyces cerevisiae CCMI 885 secretes peptides that inhibit the growth of some non-Saccharomyces wine-related strains. Appl. Microbiol. Biotechnol. 2010, 86, 965–972. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Deorowicz, S.; Kokot, M.; Grabowski, S.; Debudaj-Grabysz, A. KMC 2: Fast and resource-frugal k-mer counting. Bioinformatics 2015, 31, 1569–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokot, M.; Długosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbert, M.; Rechner, S.; Müller-Hannemann, M. Gerbil: A fast and memory-efficient k-mer counter with GPU-support. Algorithms Mol. Biol. 2017, 12, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizk, G.; Lavenier, D.; Chikhi, R. DSK: K-mer counting with very low memory usage. Bioinformatics 2013, 29, 652–653. [Google Scholar] [CrossRef] [Green Version]
- Cormen, T.H.; Leiserson, C.E.; Rivest, R.L.; Stein, C. Introduction to Algorithms, 3rd ed.; MIT Press: Cambridge, MA, USA, 2009; pp. 170–180. [Google Scholar]
- Beltran, J.A.; Aguilera-Mendoza, L.; Brizuela, C.A. Optimal selection of molecular descriptors for antimicrobial peptides classification: An evolutionary feature weighting approach. BMC Genom. 2018, 19, 672. [Google Scholar] [CrossRef]
- Ghaffari, N.; Sanchez-Flores, A.; Doan, R.; Garcia-Orozco, K.D.; Chen, P.L.; Ochoa-Leyva, A.; Lopez-Zavala, A.A.; Carrasco, J.S.; Hong, C.; Brieba, L.G.; et al. Novel transcriptome assembly and improved annotation of the whiteleg shrimp (Litopenaeus vannamei), a dominant crustacean in global seafood mariculture. Sci. Rep. 2014, 4, 7081. [Google Scholar] [CrossRef]
- GO. The Go Programming Language. Available online: https://go.dev/ (accessed on 4 January 2023).
- Waghu, F.H.; Idicula-Thomas, S. Collection of antimicrobial peptides database and its derivatives: Applications and beyond. Protein Sci. 2019, 29, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinacho-Castellanos, S.A.; García-Jacas, C.R.; Gilson, M.K.; Brizuela, C.A. Alignment-Free Antimicrobial Peptide Predictors: Improving Performance by a Thorough Analysis of the Largest Available Data Set. J. Chem. Inf. Model. 2021, 61, 3141–3157. [Google Scholar] [CrossRef] [PubMed]
- Sidorczuk, K.; Gagat, P.; Pietluch, F.; Kała, J.; Rafacz, D.; Bąkała, L.; Słowik, J.; Kolenda, R.; Rödiger, S.; Fingerhut, L.C.H.W.; et al. Benchmarks in antimicrobial peptide prediction are biased due to the selection of negative data. Brief. Bioinform. 2022, 23, bbac343. [Google Scholar] [CrossRef] [PubMed]
- Reinders, J. Intel Threading Building Blocks; O’Reilly Media Inc.: Newton, MA, USA, 2007. [Google Scholar]
- Tossi, A.; Sandri, L.; Giangaspero, A. New Consensus Hydrophobicity Scale Extended to Non-Proteinogenic Amino Acids—Technische Informationsbibliothek (TIB). 2002; pp. 416–417. Available online: https://www.tib.eu/en/search/id/BLCP%3ACN054397688/New-consensus-hydrophobicity-scale-extended-to/ (accessed on 12 December 2022).
- Tomii, K.; Kanehisa, M. Analysis of amino acid indices and mutation matrices for sequence comparison and structure prediction of proteins. Protein Eng. Des. Sel. 1996, 9, 27–36. [Google Scholar] [CrossRef]
- Li, Z.R.; Lin, H.; Han, L.Y.; Jiang, L.; Chen, X.; Chen, Y.Z. PROFEAT: A web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res. 2006, 34, W32–W37. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.R.; Wallqvist, A.; Levy, R.M. Simplified amino acid alphabets for protein fold recognition and implications for folding. Protein Eng. Des. Sel. 2000, 13, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Dubchak, I.; Muchnik, I.; Holbrook, S.R.; Kim, S.H. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. USA 1995, 92, 8700–8704. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA 1984, 81, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Klein, P.; Kanehisa, M.; DeLisi, C. Prediction of protein function from sequence properties: Discriminant analysis of a data base. Biochim. Biophys. Acta (BBA) Protein Struct. Mol. Enzym. 1984, 787, 221–226. [Google Scholar] [CrossRef]
- Charton, M.; Charton, B.I. The dependence of the Chou-Fasman parameters on amino acid side chain structure. J. Theor. Biol. 1983, 102, 121–134. [Google Scholar] [CrossRef]
- Kuhn, L.A.; Swanson, C.A.; Pique, M.E.; Tainer, J.; Getzoff, E.D. Atomic and residue hydrophilicity in the context of folded protein structures. Proteins: Struct. Funct. Bioinform. 1995, 23, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Cid, H.; Bunster, M.; Canales, M.; Gazitúa, F. Hydrophobicity and structural classes in proteins. Protein Eng. Des. Sel. 1992, 5, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, P.; Ponnuswamy, P.K. Hydrophobic character of amino acid residues in globular proteins. Nature 1978, 275, 673–674. [Google Scholar] [CrossRef]
- Ponnuswamy, P.; Prabhakaran, M.; Manavalan, P. Hydrophobic packing and spatial arrangement of amino acid residues in globular proteins. Biochim. Biophys. Acta (BBA) Protein Struct. 1980, 623, 301–316. [Google Scholar] [CrossRef]
- Prabhakaran, M. The distribution of physical, chemical and conformational properties in signal and nascent peptides. Biochem. J. 1990, 269, 691–696. [Google Scholar] [CrossRef] [Green Version]
- Sweet, R.M.; Eisenberg, D. Correlation of sequence hydrophobicities measures similarity in three-dimensional protein structure. J. Mol. Biol. 1983, 171, 479–488. [Google Scholar] [CrossRef]
- Zimmerman, J.; Eliezer, N.; Simha, R. The characterization of amino acid sequences in proteins by statistical methods. J. Theor. Biol. 1968, 21, 170–201. [Google Scholar] [CrossRef]
- Wolfenden, R.V.; Cullis, P.M.; Southgate, C.C.F. Water, Protein Folding, and the Genetic Code. Science 1979, 206, 575–577. [Google Scholar] [CrossRef]
- Casari, G.; Sippl, M.J. Structure-derived hydrophobic potential: Hydrophobic potential derived from X-ray structures of globular proteins is able to identify native folds. J. Mol. Biol. 1992, 224, 725–732. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Longevity | Order | Species | # Proteins | Proteome Length | # AMP | AMP/Protein Length (%) |
|---|---|---|---|---|---|---|
| High | Chiroptera | Myotis lucifugus | 33,415 | 20,196,147 | 21,596 | 0.1069 |
| High | Rodentia | Heterocephalus glaber | 32,702 | 20,603,935 | 22,221 | 0.1078 |
| High | Chiroptera | Desmodus rotundus | 25,076 | 15,885,570 | 20,017 | 0.1260 |
| High | Chiroptera | Eptesicus fuscus | 37,833 | 26,288,993 | 21,689 | 0.0825 |
| Medium | Rodentia | Cavia porcellus | 28,662 | 18,152,925 | 20,838 | 0.1147 |
| Low | Rodentia | Rattus norvegicus | 34,707 | 23,307,820 | 14,078 | 0.0604 |
| Low | Soricomorpha | Condylura cristata | 23,989 | 15,175,763 | 19,986 | 0.1316 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carballo, G.M.; Vázquez, K.G.; García-González, L.A.; Rio, G.D.; Brizuela, C.A. Embedded-AMP: A Multi-Thread Computational Method for the Systematic Identification of Antimicrobial Peptides Embedded in Proteome Sequences. Antibiotics 2023, 12, 139. https://doi.org/10.3390/antibiotics12010139
Carballo GM, Vázquez KG, García-González LA, Rio GD, Brizuela CA. Embedded-AMP: A Multi-Thread Computational Method for the Systematic Identification of Antimicrobial Peptides Embedded in Proteome Sequences. Antibiotics. 2023; 12(1):139. https://doi.org/10.3390/antibiotics12010139
Chicago/Turabian StyleCarballo, Germán Meléndrez, Karen Guerrero Vázquez, Luis A. García-González, Gabriel Del Rio, and Carlos A. Brizuela. 2023. "Embedded-AMP: A Multi-Thread Computational Method for the Systematic Identification of Antimicrobial Peptides Embedded in Proteome Sequences" Antibiotics 12, no. 1: 139. https://doi.org/10.3390/antibiotics12010139
APA StyleCarballo, G. M., Vázquez, K. G., García-González, L. A., Rio, G. D., & Brizuela, C. A. (2023). Embedded-AMP: A Multi-Thread Computational Method for the Systematic Identification of Antimicrobial Peptides Embedded in Proteome Sequences. Antibiotics, 12(1), 139. https://doi.org/10.3390/antibiotics12010139