Abstract
The synthesis and antiribosomal and antibacterial activity of both anomers of a novel apralog, 5-O-(5-amino-3-C-dimethylaminopropyl-D-ribofuranosyl)apramycin, are reported. Both anomers show excellent activity for the inhibition of bacterial ribosomes and that of MRSA and various wild-type Gram negative pathogens. The new compounds retain activity in the presence of the aminoglycoside phosphoryltransferase aminoglycoside modifying enzymes that act on the primary hydroxy group of typical 4,5-(2-deoxystreptamine)-type aminoglycoside and related apramycin derivatives. Unexpectedly, the two anomers have comparable activity both for the inhibition of bacterial ribosomes and of the various bacterial strains tested.
1. Introduction
In our quest to improve the antibacterial activity of apramycin 1, an atypical 2-deoxystreptamine-type aminoglycoside antibiotic with reduced toxicity, minimal susceptibility to aminoglycoside modifying enzymes [1,2,3,4] (AMEs) and ribosomal methyltransferases (RMTs), and strong activity against a broad spectrum of ESKAPE pathogens [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19], we have developed the 5-O-furanosyl apramycins, or apralogs [20,21,22,23,24]. The present optimal apralogs carry aminoalkyl substituents at the 3-position of the furanosyl ring, eg, 2, and/or aminodeoxy substitution at the 5-position of the furanose ring as in 3 and 4, and have increased levels of activity against ESKAPE pathogens while retaining the outstanding toxicity profile and minimal susceptibility to resistance mechanisms that characterize apramycin itself. In our continuing quest to further improve the apralogs we designed and report here on the synthesis and evaluation of the new apralogs 5 and 6. Like the previous apralogs 2 and 4, these novel derivatives carry activity-enhancing aminoalkyl substituents at the ribose 3-position, but now appended via a carbon-carbon bond as opposed to the previous ether linkages. This modification allows the retention of a hydroxy group at the ribose 3-position with the potential to engage in adventitious hydrogen bonding interactions in the hydrated binding site and the consequent potential to further increase activity and selectivity (Figure 1). Ultimately, we find that 5 and 6 have essentially identical activity and ribosomal selectivity, indicating that the modifications introduced override the importance of anomeric configuration in the ribofuranosyl bond that characterized the early apralogs.
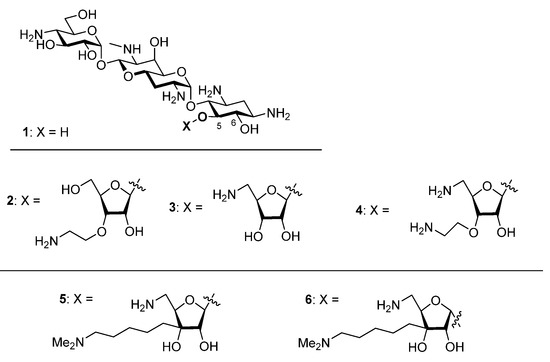
Figure 1.
Apramycin 1, Apralogs 2–4, and the Target Compounds 5 and 6.
2. Results
2.1. Synthesis
Apralogs 5 and 6 were synthesized by glycosylation of selectively protected apramycin derivative 18 with glycosyl donor 17 as the key step (Scheme 1). Alcohol 18 was accessed from apramycin 1 in four steps as described previously [25,26], whereas 3C-aminoalkyl ribofuranose 17 was synthesized from protected xylofuranose 7 (Scheme 1). Thus, treatment of 7 with thionyl chloride in pyridine furnished cyclic sulfite [27] that was further reacted with NaN3 to afford 8 in 86% yield. Subsequent oxidation with Dess-Martin periodinane (DMP) delivered ketone 9 that underwent highly stereoselective addition of Grignard reagent 11, prepared from THP-protected bromopentanol 10 [28] and metallic magnesium. Subsequent benzoyl protection of the resulting tertiary alcohol was followed by THP cleavage with Bu4N-Br3 and oxidation of the so-formed primary alcohol to aldehyde 15 in 86% yield over three steps from 12. The desired N,N-dimethylamino moiety was installed by reductive amination of 15 in 72% yield, after which a swap of the acetonide protection for the corresponding 2,3-diacetate delivered glycosyl donor 17 in 83% yield as a 3:2 mixture of α:β-anomers.
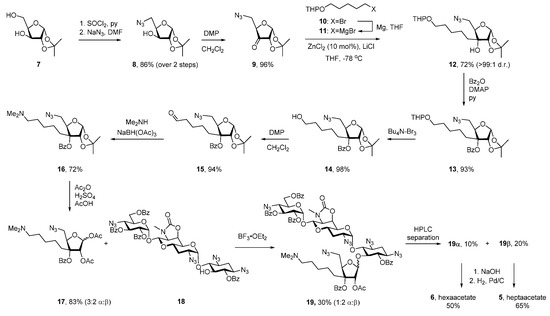
Scheme 1.
Synthesis of Apralogs 5 and 6.
Glycosidic bond formation between glycosyl donor 17 and alcohol 18 was a non-trivial task. Initial attempts using excess (6 equiv) of BF3 OEt2, TMS-OTf and TES-OTf as acidic promoters in the presence of 3Å MS (type A zeolite, 400 mg per mmol of 17) as water scavenger only led to 5% conversion of alcohol 18, perhaps because of the alkaline nature of zeolite sieves [29]. While pretreatment of the molecular sieves with acid resulted in a slight improvement of the BF3 OEt2-promoted glycosylation (10%), a preparative useful 30% yield of the desired 19 was eventually obtained in the absence of molecular sieves. Under these conditions glycoside 19 was formed as 1:2 mixture of α:β-anomers that were obtained as individual isomers after straightforward separation from unreacted 18 and hydrolyzed glycosyl donor 17 by preparative HPLC. Each of epimeric glycosides 19β and 19α was deprotected by a sequence of saponification, followed by hydrogenolysis of azides (Scheme 1), with final purification achieved by preparative HPLC, followed by treatment with acetic acid and trituration with MeCN to give apralogs 5 and 6 in the form of their peracetate salts.
2.2. Activity and Selectivity at the Drug Target
We first checked the activity of the new apralogs for activity at the target level, the ribosomal decoding A site [30,31,32,33,34,35,36], through their ability to disrupt bacterial protein synthesis in cell-free translation assays [37], with apramycin 1 and the apralogs 2, 3 and 4 as comparators (Table 1). We also screened for inhibition of protein synthesis by a set of humanized bacterial ribosomes in which the complete bacterial decoding A site has been replaced by that of the human mitochondrial (Mit13) or A1555G mutant mitochondrial ribosome (A1555G) (Figure 2) [38], as AGA binding to the cognate decoding A sites of the human mitochondrial and especially the A1555G mutant mitochondrial ribosomes in the cochlea is considered to be one of the main causes of AGA-induced ototoxicity [30,39,40,41,42,43,44,45]. Finally, we screened for inhibition of protein synthesis by similarly engineered bacterial ribosomes carrying the human cytosolic decoding A site (Cyt14) to assess the possibility of broader systemic toxicity (Figure 2).

Table 1.
Antiribosomal Activities and Selectivities (IC50, µM) a.
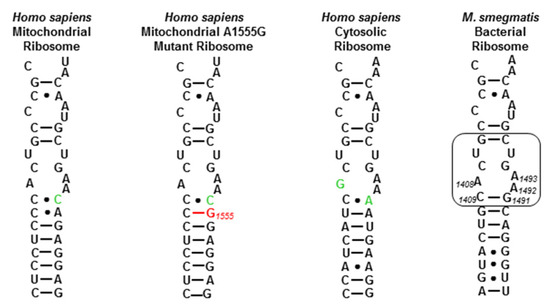
Figure 2.
Decoding A sites of prokaryotic and eukaryotic ribosomes. The bacterial AGA binding pocket is boxed. The bacterial numbering scheme is illustrated for the AGA binding pocket. Changes from the bacterial ribosome binding pocket are colored green. The A1555G mutant conferring hypersusceptibility to AGA ototoxicity is colored red.
Compounds 5 and 6 show very similar levels of activity for the inhibition of the wild-type bacterial ribosome and for that of the hybrid ribosomes carrying the eukaryotic decoding A sites, indicating that the anomeric configuration of the ribofuranosyl ring is of no consequence in this pair of isomers. The activity of 5 and 6 against the wild-type bacterial ribosome is comparable to that of 2, 2-fold better than of apramycin itself and the apralog 3 and 2–3-fold-less than that of apralog 4. In terms of selectivity for the bacterial ribosome over the three eukaryotic hybrid ribosomes, the two novel compounds retain the overall favorable profile of apramycin and the apralogs in general (Table 1).
2.3. Antibacterial Activity against Wild-Type Bacterial Strains
All newly prepared compounds and the comparators were tested for activity against a series of ESKAPE pathogens made up of a Gram-positive methicillin-resistant Staphylococcus aureus (MRSA) strain, and a panel of wild-type Gram negative pathogens (Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Acinetobacter baumannii, Pseudomonas aeruginosa) (Table 2).
Table 2.
Antibacterial Activities against Wild-Type E. coli and ESKAPE Pathogens (MIC, μg/mL) a.
Consonant with their inhibition of the wild-type bacterial ribosomes, compounds 5 and 6 have very similar antibacterial activity against MRSA and the wild-type Gram negative pathogens screened (Table 2). Again in agreement with the antiribosomal activities, the two compounds display comparable activity to apramycin itself and to the apralogs 2 and 3, and 2-fold less activity than 4 against all pathogens tested, with the exceptions of Acinetobacter baumannii and Pseudomonas aeruginosa, where they showed 4–8 fold less activity.
2.4. Antibacterial Activity against Resistant Bacterial Strains
To gauge the ability of the new apralogs to overcome resistance due to the presence of AMEs, they were screened against a panel of engineered E. coli each member of which carries a specific resistance determinant (Table 3). Four APH isoforms were included in this survey, together with one bearing the AAC(3)-IV AME known to be problematic in the apramycin series [20,21], and two carrying G1405-acting RMTs (ArmA and RmtB), which strongly mitigate the activity of all DOS-type AGAs currently used in the clinic (Table 3).
Table 3.
Activities against E. coli in the Presence of Specific Resistance Determinants (MIC, μg/mL) a.
As indicated in Table 3, 5 and 6 retained excellent activity against E. coli strains bearing four different APH(3′) isoforms and in particular against the APH(3′,5′′)-Ia isoform [46], which has the ability to phosphorylate at the ribose 5-position and so abrogate the activity of the 4,5-DOS AGAs in general and of apralogs such as 2 that retain the hydroxy group in the ribose side chain. Notably, like other apralogs, 5 and 6 afford a significant measure of protection against the action of the AAC(3)-IV isozyme, the only AME with the ability to modify and reduce the activity of apramycin itself [47]. Finally, the novel modification in 5 and 6 does not lead to resistance arising from the presence of ribosomal methyltransferases acting on G1405 [48].
2.5. Discussion
Compounds 5 and 6 retain excellent levels of activity for inhibition of the bacterial ribosome and correspondingly strong levels of antibacterial activity against MRSA and wild-type Gram negative pathogens. Compounds 5 and 6 show comparable selectivity for inhibition of the bacterial ribosome over the eukaryotic ribosomes to other apralogs and a similar profile to other 5′′-amino-5′′-deoxy apralogs when challenged with E. coli carrying the APH(3′)-Ia and AAC(3)-IV AMEs. As such the novel 3-C-(aminoalkyl)-3-hydroxy modification in the ribose ring of the apralogs is a viable modification, but based on the present data does not offer any particular advantages over the existing series of compounds and in particular the advanced apralog 4. It is, however, noteworthy, that the antiribosomal and antibacterial activities of the two compounds are essentially identical, indicating that the anomeric configuration in the ribofuranosyl ring is of no consequence in this series. This observation differs significantly from that previously reported for 2 and its α-ribofuranosyl epimer 20 (Figure 3), where the β-isomer was some 400 times more active for inhibition of the bacterial ribosome, and between 2- and 8-fold more active in MIC assays against wild-type Gram negative organisms [20]. As the β-ribofuranosyl configuration is usually necessary to position the primary side chain hydroxyl group of the ribose moiety for a critical hydrogen bonding interaction with both N2′ in ring I and with G1491 in the drug binding pocket, this result suggests that the N2′-OH/NH25′’-G1491 hydrogen bond is not critical in the present molecules. This is presumably because of the presence of six basic amines, which we have previously shown surmounts the importance of this hydrogen bond [22].
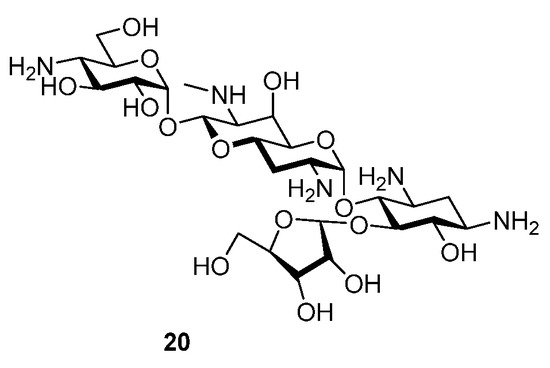
Figure 3.
Structure of Apralog 20.
3. Conclusions
The synthesis of the α- and β-anomers of a novel 5-O-(3C-aminoalkyl-5-aminoribofuranosyl)apramycin is described. The new modification affords strong activity for the inhibition of protein synthesis by the bacterial ribosome, and for the inhibition of MRSA and typical Gram-negative pathogens. Consistent with other apralogs carrying the 5-amino-5-deoxy modification in the ribofuranosyl ring, the new compounds are not susceptible to deactivation by the APH(3′,5′′)-Ia type AME. Unexpectedly, both anomers of the new compound show essentially identical activity.
4. Materials and Methods
4.1. General Experimental
All reagents and solvents were purchased from commercial suppliers and were used without further purification unless otherwise specified. All experiments were carried out under a dry argon atmosphere unless otherwise specified. Unless noted otherwise, progress of reactions was monitored by thin-layer chromatography on pre-coated aluminum-backed silica gel plates (Merck Kieselgel 60F254, Merck, Darmstadt, Germany) and were visualized by UV light (254 nm) and by charring with sulfuric acid in ethanol (20:80, v/v), or potassium permanganate solution [preparation: 1.5 g of KMnO4, 10 g of K2CO3, 1.25 mL of 10% sol. of NaOH in 200 mL of H2O], or vanillin solution [preparation: 15 g of vanillin in 250 mL of ethanol and 2.5 mL of conc. H2SO4]. Flash column chromatography was performed using an IsoleraTM automated flash purification system (Biotage AB, Uppsala, Sweden) equipped with KP-Sil 10–100 g flash cartridges (Biotage AB, Uppsala, Sweden) for normal phase separations and C18 25 μm flash cartridges (Biotage AB, Uppsala, Sweden) for reverse phase separations. Optical rotations were measured at 589 nm and 20 °C on a digital polarimeter with a path length of 10 cm. 1H and 13C NMR spectra of all compounds were recorded using at 400 MHz and 600 MHz instruments unless otherwise specified and assignments made with the help of COSY, HMBC, and HSQC spectra. ESI-HRMS were recorded using a time-of-flight mass spectrometer fitted with an electrospray source. Copies of 1H and 13C NMR spectra for all new compounds are provided in the Supplementary Material.
4.2. 5-Azido-5-deoxy-1,2-O-isopropylidene-α-D-xylofuranose (8)
The title compound was prepared according to literature procedure [49]. Accordingly, a stirred solution of 1,2-O-isopropylidine-α-D-xylofuranose 7 (5 g, 26.29 mmol, 1 equiv) in anhydrous dichloromethane (100 mL) was cooled to 0 °C (crushed ice bath) and treated with anhydrous pyridine (4.89 mL, 60.46 mmol, 2.3 equiv) under argon atmosphere. Then, a solution of SOCl2 (2.19 mL, 30.23 mmol, 1.15 equiv) in anhydrous dichloromethane (20 mL) was added dropwise at 0 °C over a period of 20 min. The resulting yellowish solution was stirred at 0 °C for 2 h, and the reaction progress was monitored by GC-MS assay. Upon completion of the reaction, a solution was transferred to a separatory funnel and washed with water (3 × 50 mL). The DCM layer was dried over Na2SO4, filtered off concentrated under reduced pressure keeping the water bath temperature below 30 °C to avoid product decomposition. The yellow residue was dissolved in anhydrous DMF (50 mL) and NaN3 (5.12 g, 78.9 mmol, 3 equiv) was added. The resulting brown suspension was heated at 110 °C with stirring for 18 h, then it was cooled to ambient temperature and all volatiles were removed in vacuo. The residue was dissolved in EtOAc (100 mL) and washed with water (100 mL). The water layer was back-extracted with Et2O (3 × 100 mL). The combined EtOAc and Et2O extracts were washed with water (100 mL) to remove residual DMF and inorganic salts, then dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting yellow oily residue was purified on Biotage SNAP KP-Sil 50 g silica cartridge (gradient elution from 100% petroleum ether (PE) to 45% EtOAc/PE) to give 8 (4.85 g, 86%) as a colorless sticky mass. 1H NMR (400 MHz, CDCl3, ppm) δ 5.95 (d, J = 3.7 Hz, 1H), 4.52 (d, J = 3.7 Hz, 1H), 4.31–4.23 (m, 2H), 3.66–3.57 (m, 2H), 2.22 (d, J = 5.2 Hz, 1H), 1.50 (s, 3H), 1.32 (d, J = 0.8 Hz, 3H). The 1H NMR spectrum was in agreement with that reported in the literature [50].
4.3. 5-Azido-5-deoxy-1,2-O-isopropylidene-α-D-erythro-pentofuranos-3-ulose (9)
A stirred colorless solution of xylofuranose 8 (0.80 g, 3.72 mmol) in anhydrous dichloromethane (10 mL) was cooled to 0 °C (crushed ice bath), and Dess-Martin periodinane (2.05 g, 4.83 mmol, 1.3 equiv) was added under argon atmosphere. After stirring at 0 °C for 20 min, the white suspension was warmed to ambient temperature and stirred for additional 2 h. The reaction progress was monitored by GC-MS assay. Upon completion of the reaction, the yellowish suspension was diluted with 10% aqueous sodium thiosulfate solution (30 mL) and transferred to a separation funnel. Layers were separated and the organic layer was washed with saturated aqueous NaHCO3 solution (50 mL), brine, dried over Na2SO4 and filtered. Concentration under reduced pressure afforded yellowish residue that was purified on Biotage SNAP KP-Sil 25 g silica cartridge (gradient elution from 100% PE to 40% EtOAc/PE) to give 9 (0.76 g, 96%) as a colorless oil. 1H NMR (400 MHz, CDCl3, ppm) δ 6.15 (d, J = 4.4 Hz 1H), 4.50 (td, J = 3.3, 1.1 Hz, 1H), 4.38 (dd, J = 4.4, 1.1 Hz, 1H), 3.68 (dd, J = 13.2, 3.3 Hz, 1H), 3.54 (dd, J = 13.2, 3.3 Hz, 1H), 1.49 (s, 3H), 1.43 (s, 3H). The 1H NMR spectrum was in agreement with that reported in the literature [51].
4.4. 2-(5-Bromopentyloxy)-tetrahydro-2H-pyran (10)
To a stirred solution of 5-bromo-1-pentanol (7.0 mL, 57.83 mmol) in anhydrous DCM (75 mL) was added p-toluenesulfonic acid hydrate (1.10 g, 5.78 mmol, 0.1 equiv) under argon atmosphere. The resulting clear solution was cooled to 0 °C (crushed ice bath) and 3,4-dihydro-2H-pyran (7.9 mL, 86.74 mmol, 1.5 equiv) was added dropwise over a period of 20 min. The resulting colorless solution was warmed to ambient temperature and stirred for 18 h. The reaction progress was monitored by GC-MS assay. After complete conversion, the reaction mixture was diluted with water (100 mL), layers were separated and the aqueous layer was back-extracted with DCM (3 × 50 mL). The combined DCM extracts were washed with brine (100 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The yellowish oily residue was purified on Biotage SNAP KP-Sil 100 g silica cartridge (gradient elution from 100% PE to 5% EtOAc/PE) to give 10 (12.74 g, 88%) as a colorless oil. 1H NMR (400 MHz, CDCl3, ppm) δ 4.64–4.52 (m, 1H), 3.91–3.83 (m, 1H), 3.79–3.71 (m, 1H), 3.53–3.47 (m, 1H), 3.45–3.36 (m, 3H), 1.93–1.79 (m, 3H), 1.76–1.47 (m, 9H). The 1H NMR spectrum was in agreement with that reported in the literature [28].
4.5. (5-((Tetrahydro-2H-pyran-2-yl)oxy)pentyl)magnesium bromide (11)
An oven-dried round-bottom two neck flask equipped with magnetic stir-bar was cooled to ambient temperature under an argon atmosphere. Magnesium turnings (2.66 g, 101.13 mmol, 2 equiv) were placed in the flask and activated by intensive stirring for 12 h under argon atmosphere at ambient temperature. Anhydrous THF (5 mL) was then added, a reflux condenser was mounted and the slurry was heated at 60 °C (water bath) under an argon atmosphere. 1,2-Dibromomethane (435 µL, 0.1 equiv) was added dropwise under argon, and after gas evolution ceased, a solution of bromide 10 (12.74 g, 50.57 mmol, 1 equiv) in anhydrous THF (50 mL) was added dropwise at 60 °C over a period of 45 min. The resulting gray suspension was stirred at ambient temperature for additional 3 h, then stirring was turned off and the suspension was left undisturbed overnight under argon atmosphere. The supernatant was carefully transferred via cannula to an oven-dried round-bottom flask and diluted with anhydrous THF (45 mL). Concentration of the Grignard reagent 11 was determined to be 0.38 M by titration with menthol and 1,10-phenanthroline [52].
4.6. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(6-(5-((tetrahydro-2H-pyran-2-yl)oxy)pentyl)-α-D-ribofuranose (12)
Grignard reagent 11 (0.38 M solution in THF, 34.0 mL, 13.6 mmol, 2 equiv), ZnCl2 (0.7 M solution in anhydrous THF, 3.9 mL, 2.7 mmol, 0.4 equiv) and LiCl (0.5 M solution in anhydrous THF, 27.2 mL, 13.6 mmol, 2 equiv) were mixed and the resulting gray solution was stirred at ambient temperature for 30 min, whereupon it was cooled to −78 °C (dry ice/acetone bath). A solution of ketone 9 (1.45 g, 6.80 mmol) in anhydrous THF (2.5 mL) was added rapidly at a rate to keep temperature below −60 °C. The resulting yellow suspension was stirred at −78 °C for 1 h, warmed to ambient temperature over a period of 30 min and quenched with saturated aqueous NH4Cl solution (25 mL). The yellow slurry was transferred to a separation funnel, diluted with water (100 mL), and the product was back-extracted with EtOAc (3 × 50 mL). The organic extracts were combined, dried over Na2SO4 and filtered off. The solvent was evaporated under reduced pressure and the yellow residue was purified on a KP-Sil 50 g silica cartridge (gradient elution from 10% EtOAc/PE to 50% EtOAc/PE) to give 12 (1.88 g, 72%) as a yellowish viscous oil; analytical TLC on silica gel, 1:1 EtOAc/PE, Rf = 0.60. +21.5 (c 0.40, CHCl3). 1H NMR (400 MHz, CDCl3, ppm) δ 5.78 (d, J = 3.9 Hz, 1H), 4.59–4.55 (m, 1H), 4.31 (d, J = 3.9 Hz, 1H), 3.90 (dd, J = 7.0, 4.8 Hz, 1H), 3.85 (dt, J = 7.5, 3.6 Hz, 1H), 3.74 (dtd, J = 9.2, 7.0, 2.4 Hz, 1H), 3.53–3.46 (m, 1H), 3.45–3.35 (m, 3H), 2.61 (s, 1H), 1.88–1.78 (m, 1H), 1.76–1.69 (m, 1H), 1.64–1.48 (m, 8H), 1.58 (s, 3H), 1.45–1.36 (m, 4H), 1.37 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3, ppm) δ 112.7, 103.8, 99.0, 81.7, 80.5, 79.0, 67.6, 62.5, 49.7, 30.9, 30.7, 29.8, 27.0, 26.7, 26.6, 25.6, 22.9, 19.8. HRMS (ESI/Q-TOF) m/z: [M-acetone + H]+ Calculated C15H26N3O5: 328.3848. Found: 328.3822.
4.7. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(6-(5-((tetrahydro-2H-pyran-2-yl)oxy)pentyl)-3-O-benzoyl-α-D-ribofuranose (13)
Benzoic anhydride (2.13 g, 9.42 mmol, 3 equiv) and DMAP (192 mg, 1.57 mmol, 0.5 equiv) were added to a stirred solution of tertiary alcohol 12 (1.21 g, 3.12 mmol) in anhydrous pyridine (15 mL) at 0 °C (crushed ice bath). The resulting yellowish solution was heated at 100 °C for 18 h. After cooling to ambient temperature, volatiles were evaporated under reduced pressure. The yellow residue was diluted with EtOAc (50 mL) and washed with saturated aqueous NaHCO3 solution (3 × 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The yellow oily residue was purified on KP-Sil 50 g silica cartridge (gradient elution from 10% EtOAc/PE to 50% EtOAc/PE) to give 13 (1.43 g, 93%) as a yellowish sticky mass; analytical TLC on silica gel, 1:1 EtOAc/PE, Rf = 0.63. +22.8 (c 0.57, CHCl3).1H NMR (400 MHz, CDCl3, ppm) δ 8.04–7.98 (m, 2H), 7.62–7.55 (m, 1H), 7.49–7.42 (m, 2H), 5.81 (d, J = 3.7 Hz, 1H), 4.94 (d, J = 3.7 Hz, 1H), 4.55–4.49 (m, 1H), 4.37 (dd, J = 7.2, 4.8 Hz, 1H), 3.82 (ddd, J = 11.1, 7.6, 3.3 Hz, 1H), 3.74–3.65 (m, 1H), 3.63–3.57 (m, 2H), 3.47 (dt, J = 10.4, 5.0 Hz, 1H), 3.34 (dt, J = 9.5, 6.3 Hz, 1H), 2.03–1.87 (m, 2H), 1.83–1.64 (m, 2H), 1.60–1.46 (m, 9H), 1.45–1.29 (m, 7H).13C{1H} NMR (101 MHz, CDCl3, ppm) δ 165.0, 133.4, 130.1, 129.9, 128.6, 112.9, 104.0, 99.0, 85.3, 83.1, 80.0, 67.4, 62.5, 50.2, 30.9, 30.4, 29.5, 27.0, 26.9, 26.8, 25.6, 23.6, 19.8. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calculated C25H35N3O7Na: 512.2373. Found: 512.2394.
4.8. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(5-hydroxypentyl)-3-O-benzoyl-α-D-ribofuranose (14)
Tetrabutylammonium tribromide (80 mg, 0.17 mmol, 0.1 equiv) was added to a stirred solution of THP-protected alcohol 13 (810 mg, 1.65 mmol) in MeOH (15 mL) at ambient temperature. The resulting orange solution was stirred for 3 h, then acetone (25 mL) was added and the resulting solution was stirred for additional 15 min. After the volatiles were evaporated under reduced pressure, the orange oily residue was diluted with EtOAc (50 mL) and washed with saturated aqueous NaHCO3 solution (3 × 50 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The orange oily residue was purified on KP-Sil 50 g silica cartridge (gradient elution from 20% EtOAc/PE to 60% EtOAc/PE) to give 14 (655 mg, 98%) as a yellowish viscous oil; analytical TLC on silica gel, 1:1 EtOAc/PE, Rf = 0.30. +70.2 (c 0.38, CHCl3). 1H NMR (400 MHz, CDCl3, ppm) δ 8.03–7.98 (m, 2H), 7.62–7.55 (m, 1H), 7.49–7.42 (m, 2H), 5.81 (d, J = 3.7 Hz, 1H), 4.94 (d, J = 3.7 Hz, 1H), 4.37 (dd, J = 7.3, 4.8 Hz, 1H), 3.63–3.56 (m, 4H), 2.03–1.88 (m, 2H), 1.58–1.51 (m, 2H), 1.49 (s, 3H), 1.43–1.29 (m, 8H). 13C{1H} NMR (101 MHz, CDCl3, ppm) δ 165.0, 133.4, 130.0, 129.9, 128.6, 112.9, 104.0, 85.2, 83.1, 79.9, 62.8, 50.1, 32.4, 30.4, 26.8, 26.7, 26.3, 23.6. HRMS (ESI/Q-TOF) m/z: [M + Na]+ Calculated C20H27N3O6Na: 428.1798. Found: 428.1786.
4.9. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(5-oxopentyl)-3-O-benzoyl-α-D-ribofuranose (15)
A stirred solution of alcohol 14 (610 mg, 1.50 mmol) in anhydrous DCM (10 mL) was cooled to 0 °C (crushed ice bath) under argon atmosphere and treated with Dess-Martin periodinane (830 mg, 1.96 mmol, 1.3 equiv), followed by few drops of NEt3. After stirring at 0 °C for 20 min, the white suspension was warmed to ca. 10 °C and stirred at this temperature for additional 2–3 h. The progress of the reaction was followed by TLC and UPLC assays. After completion of the reaction, the white suspension was diluted with 10% aqueous sodium thiosulfate solution (25 mL) and layers were separated. The organic layer was washed with saturated NaHCO3 solution (50 mL), brine, dried over Na2SO4 and filtered. Removal of volatiles under reduced pressure afforded pale yellow oily residue that was purified on KP-Sil 10 g silica cartridge (gradient elution from 20% to 50% EtOAc/PE) to afford 15 (572 mg, 94%) as a colorless oil; analytical TLC on silica gel, 1:1 EtOAc/PE, Rf = 0.51. +69.1 (c 0.42, CHCl3). 1H NMR (400 MHz, CDCl3, ppm) δ 9.73 (t, J = 1.4 Hz, 1H), 8.03–7.98 (m, 2H), 7.63–7.56 (m, 1H), 7.48–7.44 (m, 2H), 5.81 (d, J = 3.7 Hz, 1H), 4.93 (d, J = 3.7 Hz, 1H), 4.36 (dd, J = 6.9, 5.1 Hz, 1H), 3.63–3.55 (m, 2H), 2.46–2.40 (m, 2H), 2.06–1.93 (m, 2H), 1.67–1.58 (m, 2H), 1.49 (s, 3H), 1.40–1.32 (m, 5H). 13C{1H} NMR (101 MHz, CDCl3, ppm) δ 201.8, 165.0, 133.5, 129.9, 129.9, 128.6, 113.0, 103.9, 85.1, 83.1, 79.8, 50.0, 43.5, 30.3, 26.8, 23.3, 22.4. HRMS (ESI/Q-TOF) m/z: [M-acetone + H]+ Calculated C17H20N3O5: 346.1403. Found: 346.1414.
4.10. 5-Azido-5-deoxy-1,2-O-isopropylidene-3-C-(5-(dimethylamino)pentyl)-3-O-benzoyl-α-D-ribofuranose (16)
To a solution of aldehyde 15 (570 mg, 1.41 mmol) in anhydrous THF (15 mL) at ambient temperature were added Me2NH (2 M solution in THF, 2.1 mL, 4.2 mmol, 3 equiv) and glacial acetic acid (81 µL, 1.41 mmol, 1 equiv). The resulting yellow solution was stirred for 1 h then cooled to 0 °C (crushed ice bath), and NaBH(OAc)3 (449 mg, 2.12 mmol, 1.5 equiv) was added in 3 portions. The yellow suspension was stirred at 0 °C for 2 h whereupon water (25 mL) and saturated aqueous NaHCO3 solution (25mL) were added. The resulting cloudy solution was extracted with EtOAc (3 × 30 mL), combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The yellow oily residue was purified on KP-Sil 10 g silica cartridge (gradient elution from 100% EtOAc to 2% NEt3 in EtOAc) to give 16 (440 mg, 72%) as a yellow oil; analytical TLC on silica gel, 30% MeOH in DCM, Rf = 0.33. +48.0 (c 0.39, CHCl3). 1H NMR (400 MHz, CDCl3, ppm) δ 8.03–7.99 (m, 2H), 7.61–7.56 (m, 1H), 7.49–7.43 (m, 2H), 5.81 (d, J = 3.7 Hz, 1H), 4.94 (d, J = 3.7 Hz, 1H), 4.36 (dd, J = 7.4, 4.8 Hz, 1H), 3.64–3.55 (m, 2H), 2.21–2.16 (m, 8H), 2.05–1.88 (m, 2H), 1.49 (s, 3H), 1.45–1.38 (m, 2H), 1.37–1.25 (m, 7H). 13C{1H} NMR (101 MHz, CDCl3, ppm) δ 165.0, 133.4, 130.1, 129.9, 128.6, 112.9, 104.0, 85.3, 83.1, 80.0, 59.7, 50.2, 45.6, 30.4, 28.0, 27.5, 26.8, 26.8, 26.7, 23.7. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calculated C22H33N4O5: 433.2451. Found: 433.2462.
4.11. 1,2-Di-O-acetyl-5-azido-5-deoxy-3-C-(5-(dimethylamino)pentyl)-3-O-benzoyl-α/β-D-ribofuranose (17)
A stirred solution of 1,2-O-isopropylidene-protected ribofuranose 16 (1.1 g, 2.54 mmol) in glacial acetic acid (20 mL) and Ac2O (9.6 mL, 102 mmol, 40 equiv) was cooled to 0 °C (crushed ice bath). Concentrated H2SO4 (68 µL, 1.27 mmol, 0.5 equiv) was added dropwise at 0 °C. The yellow solution was warmed to ambient temperature and stirred for 18 h, whereupon volatiles were removed under reduced pressure keeping the water bath temperature below 30 °C. The brown oily residue was diluted with DCM (50 mL) and water (20 mL), cooled to 0 °C (crushed ice bath) and pH of aqueous layer was adjusted to the neutral by addition of saturated aqueous NaHCO3 solution. The mixture was transferred to a separation funnel, layers were separated and the aqueous layer was extracted with EtOAc (3 × 30 mL). The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting brown oily residue was purified on KP-Sil 25 g silica cartridge (gradient elution from 100% EtOAc to 2% NEt3 in EtOAc) to give 17 (1.01 g, 83%; 3:2 α:β mixture of anomers) as a yellow oil; analytical TLC on silica gel, 5% NEt3 in EtOAc, Rf = 0.40. 1H NMR (400 MHz, CDCl3, ppm) δ 8.09–7.94 (m, 2H, both anomers), 7.64–7.57 (m, 1H, both anomers), 7.51–7.43 (m, 2H, both anomers), 6.49 (d, J = 4.6 Hz, 0.6H, major anomer), 6.23 (d, J = 2.4 Hz, 0.4H, minor anomer), 5.61 (d, J = 2.4 Hz, 0.4H, minor anomer), 5.32 (d, J = 4.6 Hz, 0.6H, major anomer), 4.85 (dd, J = 4.8, 3.5 Hz, 0.6H, major anomer), 4.74 (dd, J = 6.6, 3.3 Hz, 0.4H, minor anomer), 3.84–3.81 (m, 0.4H, minor anomer), 3.81–3.77 (m, 0.6H, major anomer), 3.60–3.56 (m, 0.6H, major anomer), 3.56–3.53 (m, 0.4H, minor anomer), 2.63–2.45 (m, 1H, both anomers), 2.42–2.30 (m, 1H, both anomers), 2.18 (s, 6H, both anomers), 2.15 (s, 1.8H, major anomer), 2.14 (s, 1.2H, minor anomer), 2.13 (s, 1.2H, minor anomer), 2.04 (s, 1.8H, major anomer), 1.91–1.73 (m, 1H, both anomers), 1.47–1.34 (m, 3H, both anomers), 1.32–1.23 (m, 4H, both anomers). 13C{1H NMR (101 MHz, CDCl3, ppm; 3:2 α:β mixture of anomers) δ 169.8, 169.7, 169.7, 169.7, 165.3, 133.8, 133.8, 130.2, 129.9, 129.9, 129.8, 129.8, 128.9, 128.8, 128.7, 98.8, 93.2, 86.8, 85.5, 84.5, 84.3, 79.3, 76.4, 58.4, 51.9, 51.0, 50.9, 43.9, 31.9, 30.5, 27.1, 25.2, 23.2, 22.8, 22.7, 21.3, 21.1, 21.0, 20.8. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calculated C23H33N4O7: 477.2349. Found: 477.2315.
4.12. 5-O-[5′′′-Azido-5′′′-deoxy-2′′′-O-acetyl-3-C-(5-(dimethylamino)pentyl)-3-O-benzoyl-α/β-D-ribofuranosyl]-6,2′′,3′′,6′′-tetra-O-benzoyl-1,3,2′,4′′-tetraazido-1,3,2′,4′′-tetra(desamino)-6′,7′-oxazolidino-apramycin trifluoroacetate (19)
A mixture of ribofuranose 17 (100 mg, 0.21 mmol) and protected apramycin 18 [20] (228 mg, 0.21 mmol, 1 equiv) were co-evaporated with anhydrous toluene (5 mL) on a rotary evaporator three times, followed by overnight vacuum drying. Anhydrous DCM (5 mL) was added under an argon atmosphere, the resulting yellow solution was cooled to −10 °C (crushed ice/NaCl bath) and BF3•OEt2 (331 µL, 1.26 mmol, 6 equiv) was added. The resulting yellow solution was stirred at 0 °C for 48 h, and the reaction progress was monitored by UPLC-MS assay. Upon complete conversion of ribofuranose 17, the reaction was quenched with NEt3 (0.3 mL, 2.10 mmol, 10 equiv) at 0 °C and the resultant mixture was diluted with EtOAc (20 mL). The organic phase was washed with aqueous saturated NaHCO3 solution (50 mL), and the aqueous layer was back-extracted with DCM (3 × 30 mL). The combined EtOAc and DCM extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The brown residue was purified by reversed-phase preparative HPLC (column: XBridge® Prep C18 5 μm OBDTM, 30 x 100 mm, Waters Corporation Ltd, Dublin, Ireland) using gradient elution from 60% MeCN in 0.1% aqueous TFA solution to 95% MeCN in 0.1% aqueous TFA solution) to give 19α (32 mg, 10%, white powder) and 19β (60 mg, 20%, white powder); analytical TLC on silica gel, 5% NEt3 in EtOAc, Rf = 0.40.
19α-anomer: +72.3 (c 0.25, CHCl3).1H NMR (400 MHz, CDCl3, ppm) δ 12.7–11.9 (br s, 1H), 8.12–8.05 (m, 4H), 8.05–8.00 (m, 2H), 8.00–7.94 (m, 2H), 7.74–7.67 (m, 2H), 7.63–7.34 (m, 15H), 6.01 (t, J = 10.1 Hz, 1H), 5.73 (d, J = 3.1 Hz, 1H), 5.71 (d, J = 3.6 Hz, 1H), 5.34 (d, J = 1.8 Hz, 1H), 5.24 (t, J = 9.8 Hz, 1H), 5.19 (d, J = 1.8 Hz, 1H), 5.17 (dd, J = 10.5, 3.6 Hz, 1H), 4.78 (d, J = 6.0 Hz, 1H), 4.75 (dd, J = 6.4, 3.6 Hz, 1H), 4.67 (dd, J = 12.0, 2.3 Hz, 1H), 4.62 (dd, J = 12.2, 4.9 Hz, 1H), 4.31 (dd, J = 7.7, 2.0 Hz, 1H), 4.13–4.03 (m, 3H), 3.99–3.93 (m, 1H), 3.89 (t, J = 10.1 Hz, 1H), 3.81 (t, J = 9.3 Hz, 1H), 3.75 (t, J = 6.3 Hz, 1H), 3.63–3.55 (m, 1H), 3.55–3.47 (m, 2H), 3.47–3.38 (m, 1H), 3.08–2.66 (m, 12H), 2.43 (dt, J = 12.8, 4.2 Hz, 1H), 2.21–2.10 (m, 1H), 1.79–1.66 (m, 3H), 1.64–1.54 (m, 4H), 1.40–1.14 (m, 6H).13C{1H} NMR (101 MHz, CDCl3, ppm) δ 169.0, 165.9, 165.7, 165.4, 164.8, 164.5, 157.0, 133.6, 133.5, 133.4, 133.3, 129.9, 129.8, 129.6, 129.5, 129.3, 129.1, 129.1, 128.8, 128.6, 128.6, 128.5, 128.4, 128.3, 105.8, 100.7, 96.2, 95.6, 83.9, 82.7, 80.5, 78.5, 75.9, 75.2, 71.8, 71.0, 70.2, 69.7, 66.4, 65.8, 63.1, 60.7, 60.1, 59.1, 58.2, 57.4, 55.4, 51.2, 42.7, 42.5, 31.2, 30.2, 29.5, 27.9, 26.2, 23.6, 22.3, 19.9. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calculated C71H76N17O21: 1502.5402. Found: 1502.5424.
19β-anomer: +61.3 (c 0.31, CHCl3).1H NMR (400 MHz, CDCl3, ppm) δ 11.2–10.9 (br s, 1H), 8.10–8.06 (m, 2H), 8.05–8.00 (m, 2H), 7.98–7.91 (m, 4H), 7.71–7.65 (m, 2H), 7.65–7.59 (m, 1H), 7.56–7.50 (m, 4H), 7.47–7.40 (m, 3H), 7.40–7.30 (m, 5H), 7.25–7.21 (m, 2H), 6.00 (t, J = 10.1 Hz, 1H), 5.75–5.67 (m, 2H), 5.43 (s, 1H), 5.30 (s, 1H), 5.24 (t, J = 9.9 Hz, 1H), 5.16 (dd, J = 10.4, 3.6 Hz, 1H), 4.80 (d, J = 5.6 Hz, 1H), 4.76 (dd, J = 6.9, 3.6 Hz, 1H), 4.68 (dd, J = 12.2, 2.4 Hz, 1H), 4.62 (dd, J = 12.2, 4.9 Hz, 1H), 4.22 (dd, J = 8.6, 2.2 Hz, 1H), 4.15–4.06 (m, 3H), 3.89 (t, J = 10.1 Hz, 1H), 3.84–3.79 (m, 1H), 3.78–3.73 (m, 2H), 3.57–3.49 (m, 3H), 3.45 (ddd, J = 12.4, 10.0, 4.8 Hz, 1H), 3.07–2.99 (m, 3H), 2.93 (s, 3H), 2.91–2.82 (m, 6H), 2.45 (dt, J = 13.0, 4.5 Hz, 1H), 2.10–1.99 (m, 1H), 1.79–1.64 (m, 7H), 1.64–1.53 (m, 2H), 1.38–1.24 (m, 4H).13C{1H} NMR (101 MHz, CDCl3, ppm) δ 169.1, 166.3, 166.1, 165.8, 165.3, 164.5, 157.4, 133.9, 133.8, 133.8, 133.7, 133.5, 130.1, 130.0, 129.9, 129.9, 129.7, 129.5, 129.0, 128.9, 128.9, 128.8, 128.7, 128.7, 128.6, 128.5, 128.4, 106.9, 100.5, 96.4, 95.9, 83.4, 83.3, 80.9, 79.3, 76.7, 75.3, 72.0, 71.4, 70.6, 70.0, 66.6, 66.0, 63.5, 61.1, 60.4, 59.3, 58.5, 58.1, 56.0, 51.3, 43.3, 43.2, 31.5, 30.4, 29.5, 28.5, 26.5, 24.1, 22.8, 20.9. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calculated C71H76N17O21: 1502.5402. Found: 1502.5435.
4.13. 5-O-[5′′′-Amino-5′′′-deoxy-3-C-(5-(dimethylamino)pentyl)-β-D-ribofuranosyl]-apramycin heptaacetate (5)
A mixture of apramycin derivative 19β (80 mg, 0.05 mmol) in dioxane (1 mL) and NaOH (2 M aqueous solution, 240 µL, 0.48 mmol, 9 equiv) was heated at 80 °C for 24 h. The reaction progress was monitored by UPLC-MS assay. Upon complete conversion, the colorless solution was cooled to 0 °C (crushed ice bath) and dry ice was added portion-wise until pH 8. The resulting solution was concentrated to dryness under reduced pressure. The white residue was dissolved in a mixture of dioxane/deionized water/glacial acetic acid (1:1:1, 3 mL) and 10% Pd on carbon (85 mg, 0.08 mmol, 1.5 equiv) was added. The black suspension was vigorously stirred under 3 atm hydrogen pressure at ambient temperature for 18 h, and the progress of the reaction was monitored by UPLC-MS assay. Upon complete conversion, the black suspension was filtered through the pad of Celite®®®, and the filter cake was washed with 1:1 AcOH:water mixture. Combined filtrates were evaporated under reduced pressure, and the sticky oil was purified by reversed-phase preparative HPLC (column: XBridge® BEH Prep OBDTM Amide, 5 μm, 30 x 100 mm, Waters Corporation Ltd, Dublin, Ireland) using gradient elution from 95:5 A:B to 10:90 A:B (eluent A: 0.1% solution of AcOH in MeCN; eluent B: 0.1% solution of AcOH in water). The product-containing fractions (identified by ESI-MS) were combined and concentrated. Glacial acetic acid was added to the sticky oily residue, and subsequent trituration with MeCN afforded the heptaacetate salt of 5 as a white amorphous solid (32 mg, 50% yield). +64.4 (c 0.104, H2O). 1H NMR (600 MHz, D2O, ppm) δ 5.72 (d, J = 3.9 Hz, 1H), 5.41 (d, J = 3.9 Hz, 1H), 5.35 (d, J = 4.3 Hz, 1H), 5.12 (d, J = 8.5 Hz, 1H), 4.49 (t, J = 2.8 Hz, 1H), 4.09–4.04 (m, 2H), 4.02 (d, J = 4.1 Hz, 1H), 3.90 (t, J = 9.3 Hz, 1H), 3.88–3.83 (m, 2H), 3.83 (t, J = 10.1 Hz, 1H), 3.79–3.72 (m, 2H), 3.69 (dd, J = 12.4, 4.5 Hz, 1H), 3.65–3.55 (m, 3H), 3.38 (ddd, J = 13.7, 9.9, 4.1 Hz, 1H), 3.28 (dd, J = 8.5, 2.8 Hz, 1H), 3.24 (td, J = 12.5, 4.2 Hz, 1H), 3.21–3.12 (m, 2H), 3.05–2.98 (m, 3H), 2.75 (s, 6H), 2.68 (s, 3H), 2.33 (dt, J = 12.5, 4.2 Hz, 1H), 2.27 (dt, J = 9.0, 4.6 Hz, 1H), 2.01–1.92 (m, 1H), 1.82 (s, 21H), 1.73 (q, J = 12.7 Hz, 1H), 1.66–1.54 (m, 3H), 1.49–1.35 (m, 2H), 1.33–1.22 (m, 3H). 13C{1H} NMR (151 MHz, D2O, ppm) δ 180.2, 106.7, 93.8, 93.0, 92.1, 82.7, 81.6, 78.4, 77.8, 73.8, 71.1, 69.6, 69.0, 68.9, 67.7, 65.3, 62.0, 59.6, 58.7, 56.9, 51.3, 49.5, 48.1, 47.0, 41.8, 39.7, 31.0, 29.3, 27.9, 26.4, 25.4, 23.2, 22.3, 21.3. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calculated C33H66N7O14: 784.4668. Found: 784.4665. Anal. Calcd for C47H93N7O28: C, 44.10; H, 7.98; N, 7.66. Found: C, 44.31; H, 7.75; N, 7.74.
4.14. 5-O-[5′′′-Amino-5′′′-deoxy-3-C-(5-(dimethylamino)pentyl)-α-D-ribofuranosyl]-apramycin hexaacetate (6)
A mixture of apramycin derivative 19α (60 mg, 0.04 mmol) in dioxane (1 mL) and NaOH (2 M aqueous solution, 180 µL, 0.36 mmol, 9 equiv) was heated at 80 °C for 24 h. The reaction progress was monitored by UPLC-MS assay. Upon complete conversion, the colorless solution was cooled to 0 °C (crushed ice bath) and dry ice was added portion-wise until pH 7–8. The resulting solution was concentrated to dryness under reduced pressure. The white residue was dissolved in a mixture of dioxane/deionized water/glacial acetic acid (1:1:1, 3 mL) and 10% Pd on carbon (64 mg, 0.06 mmol, 1.5 equiv) was added at ambient temperature. The black suspension was vigorously stirred under 3 atm hydrogen pressure at ambient temperature for 18 h, and the progress of the reaction was monitored by UPLC-MS assay. Upon complete conversion, the black suspension was filtered through the pad of CeliteTM 545 (Thermo Fischer Scientific, Waltham, MA, USA), and the filter cake was washed with 1:1 AcOH:water mixture. Combined filtrates were evaporated under reduced pressure, and the sticky oil was purified by reversed-phase preparative HPLC (column: XBridge® BEH Prep OBDTM Amide, 5 μm, 30 x 100 mm, Waters Corporation Ltd, Dublin, Ireland) using gradient elution from 95:5 A:B to 10:90 A:B (eluent A—0.1% solution of AcOH in MeCN; eluent B—0.1% solution of AcOH in water). The product-containing fractions (identified by ESI-MS) were combined and concentrated. Glacial acetic acid was added to the sticky oily residue, and subsequent trituration with MeCN afforded the hexaacetate salt of 6 as a white amorphous solid (31 mg, 65% yield). +76.9 (c 0.25, H2O). 1H NMR (600 MHz, D2O, ppm) δ 5.60 (d, J = 3.5 Hz, 1H), 5.49 (d, J = 3.9 Hz, 1H), 5.45 (d, J = 4.6 Hz, 1H), 5.18 (d, J = 8.5 Hz, 1H), 4.53 (t, J = 2.8 Hz, 1H), 4.16 (dd, J = 11.4, 2.5 Hz, 1H), 4.07 (d, J = 4.5 Hz, 1H), 3.95–3.82 (m, 6H), 3.81–3.75 (m, 3H), 3.71 (dd, J = 9.8, 3.9 Hz, 1H), 3.50 (dt, J = 12.5, 4.2 Hz, 1H), 3.33–3.17 (m, 4H), 3.15–3.07 (m, 4H), 2.88 (s, 6H), 2.74 (s, 3H), 2.35–2.26 (m, 2H), 2.01–1.93 (m, 1H), 1.92 (s, 18H), 1.79–1.70 (m, 2H), 1.70–1.59 (m, 2H), 1.58–1.47 (m, 2H), 1.43–1.36 (m, 3H). 13C{1H} NMR (151 MHz, D2O, ppm) δ 181.3, 107.5, 95.0, 94.5, 93.2, 83.4, 82.3, 78.0, 72.4, 70.6, 70.4, 69.7, 66.5, 66.3, 63.0, 60.5, 59.8, 57.6, 52.1, 50.5, 49.0, 48.1, 42.5, 40.5, 31.8, 30.5, 30.3, 28.2, 26.0, 23.8, 23.2, 22.0. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calculated C33H66N7O14: 784.4668. Found: 784.4676. Anal. Calcd for C45H89N7O26: C, 44.18; H, 8.06; N, 8.01. Found: C, 44.07; H, 7.71; N, 7.69.
4.15. Cell-Free Luciferase Translation Assays
Cell-free in vitro translation inhibition assays were performed using luciferase mRNA and bacterial S30 extracts containing either wild-type bacterial or human hybrid ribosomes. In brief, firefly luciferase mRNA was transcribed in vitro using T7 RNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA) using a plasmid as template in which the mammalian promoter in pGL4.14 has been replaced by theT7 bacteriophage promoter (Promega, USA). Test articles in aqueous solution containing 0.3% Tween20 were dispensed into white 96-well plates (Eppendorf, Germany) using the TECAN D300e digital dispenser (Tecan, Switzerland). The test article dispensing volume was balanced to a total of 1.5 µL by 0.3% Tween20 in water. The reaction volume was brought to 15 µL by addition of 13.5 µL Translation Master Mix comprised of bacterial S30 extract, 0.2 mM amino acid mix, 6 µg tRNA (Sigma-Aldrich, USA), 0.4 µg hFluc mRNA, 0.3 µL protease inhibitor (cOmplete, EDTA-free, Roche, USA), 12 U RNAse inhibitor (Ribolock, Thermo Fisher Scientific, Waltham, MA, USA), and 6 µL S30 premix without amino acids (Promega, USA). Dispensing and mixing of reagents was performed on ice prior to incubating the sealed plates at 37 °C. After 1 h of incubation, the reaction was stopped on ice and 75 µL of luciferase assay reagent (Promega, USA) was added to each well. Luminescence was recorded with a plate reader (BIO-TEK FLx800, Witec AG, Littau, Switzerland).
4.16. Antibacterial Inhibition Assays
The minimal inhibitory concentrations (MIC) of synthesized compounds were determined by broth microdilution assays according to CLSI reference methodology M07 [53] as described previously [6] and using strains described previously [54]. Clinical bacterial isolates were obtained from the diagnostic laboratories of the Institute of Medical Microbiology, University of Zurich.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/antibiotics12010025/s1, Copies of 1H and 13C NMR spectra for all new compounds.
Author Contributions
Conceptualization, D.L., A.V., E.S. and D.C.; methodology, D.L., K.H., S.N.H., E.S. and D.C; writing—review and editing, D.L., S.N.H., E.S. and D.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by ERDF (Post-Doc Latvia, project No.1.1.1.2/VIAA/3/19/586).
Data Availability Statement
All supporting spectral data may be found in the Supplementary Materials.
Acknowledgments
The authors thank Erik C. Böttger, IMM, UZH, for helpful discussion.
Conflicts of Interest
S.N.H., A.V., and D.C. are cofounders of and equity holders in Juvabis AG, a biotech start-up working in the area of aminoglycoside development. All other authors declare no conflict of interest.
References
- Yang, L.; Ye, X.S. Development of Aminoglycoside Antibiotics Effective against Resistant Bacterial Strains. Curr. Top. Med. Chem. 2010, 10, 1898–1926. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Blanchard, J.S. Molecular Insights into Aminoglycoside Action and Resistance. Chem. Rev. 2005, 105, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. Med. Chem. Commun. 2016, 7, 11–27. [Google Scholar] [CrossRef]
- Zárate, S.G.; De la Cruz Claure, M.L.; Benito-Arenas, R.; Revuelta, R.; Santana, A.G.; Bastida, A. Overcoming Aminoglycoside Enzymatic Resistance: Design of Novel Antibiotics and Inhibitors. Molecules 2018, 23, 284. [Google Scholar] [CrossRef]
- Matt, T.; Ng, C.L.; Lang, K.; Sha, S.-H.; Akbergenov, R.; Shcherbakov, D.; Meyer, M.; Duscha, S.; Xie, J.; Dubbaka, S.R.; et al. Dissociation of Antibacterial Activity and Aminoglycoside Ototoxicity in the 4-Monosubstituted 2-Deoxystreptamine Apramycin. Proc. Natl. Acad. Sci. USA 2012, 109, 10984–10989. [Google Scholar] [CrossRef]
- Juhas, M.; Widlake, E.; Teo, J.; Huseby, D.L.; Tyrrell, J.M.; Polikanov, Y.; Ercan, O.; Petersson, A.; Cao, S.; Aboklaish, A.F.; et al. In-vitro Activity of Apramycin against Multidrug-, Carbapenem-, and Aminoglycoside-Resistant Enterobacteriaceae and Acinetobacter baumannii. J. Antimicrob. Chemother. 2019, 74, 944–952. [Google Scholar] [CrossRef]
- Becker, K.; Cao, S.; Nilsson, A.; Erlandsson, M.; Hotop, S.-K.; Kuka, J.; Hansen, J.; Haldimann, K.; Grinberga, S.; Fernández, T.B.; et al. Antibacterial Activity of Apramycin at Acidic pH Warrants Wide Therapeutic Window in the Treatment of Complicated Urinary Tract Infections and Acute Pyelonephritis. EBioMedicine 2021, 73, 103652–103661. [Google Scholar] [CrossRef]
- Becker, K.; Aranzana-Climent, V.; Cao, S.; Nilsson, A.; Shariatgorji, R.; Haldimann, K.; Platzack, B.; Hughes, D.; Andren, P.E.; Böttger, E.C.; et al. Efficacy of EBL-1003 (Apramycin) against Acinetobacter baumannii Lung Infections in Mice. Clin. Microbiol. Infect. 2021, 27, 1315–1321. [Google Scholar] [CrossRef]
- Smith, K.P.; Kirby, J.E. Evaluation of apramycin activity against carbapenem-resistant and -susceptible strains of Enterobacteriaceae. Diagn. Microbiol. Infect. Dis. 2016, 86, 439–441. [Google Scholar] [CrossRef]
- Kang, A.D.; Smith, K.P.; Eliopoulos, G.M.; Berg, A.H.; McCoy, C.; Kirby, J.E. In vitro Apramycin Activity against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Diagn. Microbiol. Infect. Dis. 2017, 88, 188–191. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, L.; Zhang, X.; Feng, Y.; Zong, Z. In vitro activity of neomycin, streptomycin, paromomycin and apramycin against carbapenem-resistant Enterobacteriaceae clinical strains. Front. Microbiol. 2017, 8, 2275–2281. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, S.Y.L.; Garneau-Tsodikova, S. Evaluation of Aminoglycoside and Carbapenem Resistance in a Collection of Drug-Resistant Pseudomonas aeruginosa Clinical Isolates. Microb. Drug Resist. 2018, 24, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.; Nafplioti, K.; Chatzikonstantinou, M.; Giamarellou, H.; Souli, M. Evaluation of apramycin activity against carbapenem-resistant Enterobacteriaceae and Acinetobacter baumannii. In Proceedings of the 28th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Madrid, Spain, 21–24 April 2018; p. p. 0096.
- Nafplioti, K.; Galani, I.; Adamou, P.; Moraitou, E.; Giannopoulou, P.; Chra, P.; Damala, M.; Vogiatzakis, E.; Trikka-Graphakos, E.; Baka, V.; et al. Epidemic Dissemination of a Carbapenem-Resistant Acinetobacter baumannii Clone Carrying armA. In Proceedings of the 28th European Congress of Clinical Microbiology and Infectious Diseases (ECCMID 2018), Madrid, Spain, 21–24 April 2018; p. p. 1105.
- Kang, A.D.; Smith, K.P.; Berg, A.H.; Truelson, K.A.; Eliopoulos, G.M.; McCoy, C.; Kirby, J.E. Efficacy of Apramycin against Multidrug-Resistant Acinetobacter baumannii in the Murine Neutropenic Thigh Model. Antimicrob. Agent. Chemother. 2018, 62, e02585-17. [Google Scholar] [CrossRef]
- Truelson, K.A.; Brennan-Krohn, T.; Smith, K.P.; Kirby, J.E. Evaluation of apramycin activity against methicillin-resistant, methicillin-sensitive, and vancomycin-intermediate Staphylococcus aureus clinical isolates. Diagn. Microbiol. Infect. Dis. 2018, 92, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Riedel, S.; Vijayakumar, D.; Berg, G.; Kang, A.D.; Smith, K.P.; Kirby, J.E. Evaluation of apramycin against spectinomycin-resistant and -susceptible strains of Neisseria gonorrhoeae. J. Antimicrob. Chemother. 2019, 74, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Krohn, T.; Kirby, J.E. Synergistic combinations and repurposed antibiotics active against the pandrug-resistant Klebsiella pneumoniae Nevada strain. Antimicrob. Agent. Chemother. 2019, 63, e01374-19. [Google Scholar] [CrossRef]
- Hao, M.; Shi, X.-M.; Lv, J.; Niu, S.; Cheng, S.; Du, H.; Yu, F.; Tang, Y.-W.; Kreiswirth, B.N.; Zhang, H.; et al. In vitro Activity of Apramycin against Carbapenem-Resistant and Hypervirulaent Klebsiella pneumoniae Isolates. Front. Microbiol. 2020, 11, 425. [Google Scholar] [CrossRef]
- Quirke, J.C.K.; Rajasekaran, P.; Sarpe, V.A.; Sonousi, A.; Osinnii, I.; Gysin, M.; Haldimann, K.; Fang, Q.-J.; Shcherbakov, D.; Hobbie, S.N.; et al. Apralogs: Apramycin 5-O-Glycosides and Ethers with Improved Antibacterial Activity and Ribosomal Selectivity and Reduced Susceptibility to the Aminoacyltranserferase (3)-IV Resistance Determinant. J. Am. Chem. Soc. 2020, 142, 530–544. [Google Scholar] [CrossRef]
- Sonousi, A.; Quirke, J.C.K.; Waduge, P.; Janusic, T.; Gysin, M.; Haldimann, K.; Xu, S.; Hobbie, S.N.; Sha, S.-H.; Schacht, J.; et al. An Advanced Apralog with Increased in-vitro and in-vivo Activity toward Gram-negative Pathogens and Reduced ex-vivo Cochleotoxicity. Chem. Med. Chem. 2021, 16, 335–339. [Google Scholar] [CrossRef]
- Quirke, J.C.K.; Sati, G.C.; Sonousi, A.; Gysin, M.; Haldimann, K.; Böttger, E.C.; Vasella, A.; Hobbie, S.N.; Crich, D. Structure-Activity Relationships for 5′’-Modifications of 4,5-Aminoglycoside Antibiotics. Chem. Med. Chem. 2022, 17, e202200120. [Google Scholar] [CrossRef]
- Abe, Y.; Nakagawa, S.; Naito, T.; Kawaguchi, H. Synthesis and Activity of 6-O-(3-Amino-3-deoxy-α-D-glucopyranosyl)- and 5-O-(β-D-ribofuranosyl)apramycins. J. Antibiot. 1981, 34, 1434–1446. [Google Scholar] [CrossRef]
- Zada, S.L.; Baruch, B.B.; Simhaev, L.; Engel, H.; Fridman, M. Chemical Modifications Reduce Auditory Cell Damage Induced by Aminoglycoside Antibiotics. J. Am. Chem. Soc. 2020, 142, 3077–3087. [Google Scholar] [CrossRef] [PubMed]
- Mandhapati, A.R.; Shcherbakov, D.; Duscha, S.; Vasella, A.; Böttger, E.C.; Crich, D. Importance of the 6′-Hydroxy Group and its Configuration for Apramycin Activity. ChemMedChem 2014, 9, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Mandhapati, A.R.; Yang, G.; Kato, T.; Shcherbakov, D.; Hobbie, S.N.; Vasella, A.; Böttger, E.C.; Crich, D. Structure-Based Design and Synthesis of Apramycin-Paromomycin Analogues. Importance of the Configuration at the 6′-Position and Differences Between the 6′-Amino and Hydroxy Series. J. Am. Chem. Soc. 2017, 139, 14611–14619. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.M.; Bols, M. On the Nature of the Electronic Effect of Multiple Hydroxyl Groups in the 6-Membered Ring—The Effects Are Additive But Steric Hindrance Plays A Role Too. Org. Biomol. Chem. 2017, 15, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Mansurova, M.; Klusák, V.; Nešněrová, P.; Muck, A.; Doubský, J.; Svatoš, A. Design and synthesis of bombykol analogues for probing pheromone-binding protein–ligand interactions. Front. Physiol. 2009, 65, 1069–1076. [Google Scholar] [CrossRef]
- Christensen, H.M.; Oscarson, S.; Jensen, H.H. Common side reactions of the glycosyl donor in chemical glycosylation. Carbohydr. Res. 2015, 408, 51–95. [Google Scholar] [CrossRef]
- Armstrong, E.S.; Kostrub, C.F.; Cass, R.T.; Moser, H.E.; Serio, A.W.; Miller, G.H. Aminoglycosides. In Antibiotic Discovery and Development; Dougherty, T.J., Pucci, M.J., Eds.; Springer Science+Business Media: New York, NY, USA, 2012; pp. 229–269. [Google Scholar]
- Böttger, E.C.; Crich, D. Aminoglycosides: Time for Resurrection of a Neglected Class of Antibacterials? ACS Infect. Dis. 2020, 6, 168–172. [Google Scholar] [CrossRef]
- Brodersen, D.E.; Clemons, W.M.; Carter, A.P.; Morgan-Warren, R.; Wimberly, B.T.; Ramakrishnan, V. The Structural Basis for the Action of the Antibiotics Tetracycline, Pactamycin, and Hygromycin B on the 30S Ribosomal Subunit. Cell 2000, 103, 1143–1154. [Google Scholar] [CrossRef]
- Moazed, D.; Noller, H.F. Interaction of Antibiotics with Functional Sites in 16S Ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef]
- François, B.; Russell, R.J.M.; Murray, J.B.; Aboul-ela, F.; Masquid, B.; Vicens, Q.; Westhof, E. Crystal Structures of Complexes Between Aminoglycosides and Decoding A Site Oligonucleotides: Role of the Number of rings and Positive Charges in the Specific Binding Leading to Miscoding. Nucleic Acids Res. 2005, 33, 5677–5690. [Google Scholar] [CrossRef] [PubMed]
- Fourmy, D.; Recht, M.I.; Blanchard, S.C.; Puglisi, J.D. Structure of the A Site of Escherichia coli 16S Ribosomal RNA Complexed with an Aminoglycoside Antibiotic. Science 1996, 274, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhou, D.; Steitz, T.A.; Polikanov, Y.S.; Gagnon, M.G. Ribosome-Targeting Antibiotics: Modes of Action, Mechanisms of Resistance, and Implications for Drug Design. Ann. Rev. Biochem. 2018, 87, 451–478. [Google Scholar] [CrossRef] [PubMed]
- Duscha, S.; Boukari, H.; Shcherbakov, D.; Salian, S.; Silva, S.; Kendall, A.; Kato, T.; Akbergenov, R.; Perez-Fernandez, D.; Bernet, B.; et al. Identification and Evaluation of Improved 4′-O-(Alkyl) 4,5-Disubstituted 2-Deoxystreptamines as Next Generation Aminoglycoside Antibiotics. mBio 2014, 5, e01827-14. [Google Scholar] [CrossRef] [PubMed]
- Hobbie, S.N.; Kalapala, S.K.; Akshay, S.; Bruell, C.; Schmidt, S.; Dabow, S.; Vasella, A.; Sander, P.; Böttger, E.C. Engineering the rRNA Decoding Site of Eukaryotic Cytosolic Ribosomes in Bacteria. Nucl. Acids Res. 2007, 35, 6086–6093. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, M.; Karasawa, T.; Steyger, P.S. Aminoglycoside-Induced Cochleotoxicity: A Review. Front. Cell. Neurosci. 2017, 11, 308. [Google Scholar] [CrossRef]
- Böttger, E.C.; Schacht, J. The Mitochondrion: A Perpetrator of Acquired Hearing Loss. Hearing Res. 2013, 303, 12–19. [Google Scholar] [CrossRef]
- Prezant, T.R.; Agapian, J.V.; Bohlman, M.C.; Bu, X.; Öztas, S.; Qiu, W.-Q.; Arnos, K.S.; Cortopassi, G.A.; Jaber, L.; Rotter, J.I.; et al. Mitochondrial Ribosomal RNA Mutation Associated with Both Antibiotic-Induced and Non-Syndromic Deafness. Nat. Genet. 1993, 4, 289–294. [Google Scholar] [CrossRef]
- Huth, M.E.; Ricci, A.J.; Cheng, A.G. Mechanisms of Aminoglycoside Ototoxicity and Targets of Hair Cell Protection. Int. J. Otolaryngol. 2011, 2011, 937861–937879. [Google Scholar] [CrossRef]
- Hobbie, S.N.; Akshay, S.; Kalapala, S.K.; Bruell, C.; Shcherbakov, D.; Böttger, E.C. Genetic Analysis of Interactions with Eukaryotic rRNA Identify the Mitoribosome as Target in Aminoglycoside Ototoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 20888–20893. [Google Scholar] [CrossRef]
- Hobbie, S.N.; Bruell, C.M.; Akshay, S.; Kalapala, S.K.; Shcherbakov, D.; Böttger, E.C. Mitochondrial Deafness Alleles Confer Misreading of the Genetic Code. Proc. Natl. Acad. Sci. USA 2008, 105, 3244–3249. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Guan, M.-X. Interaction of Aminoglycosides with Human Mitochondrial 12S rRNA Carrying the Deafness-Associated Mutation. Antimicrob. Agent. Chemother. 2009, 53, 4612–4618. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D.; Thompson, P.R. Aminoglycoside Phosphotransferases: Proteins, Structure, and Mechanism. Front. Biosci. 1999, 4, d9–d21. [Google Scholar]
- Plattner, M.; Gysin, M.; Haldimann, K.; Becker, K.; Hobbie, S.N. Epidemiologic, Phenotypic, and Structural Characterization of Aminoglycoside-Resistance Gene AAC(3)-IV. Int. J. Mol. Sci. 2020, 21, 6133. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Zhang, J.-C.; Maharjan, S.; Doumith, M.; Woodford, N. Activity of Aminoglycosides, Including ACHN-490, against Carbapenem-Resistant Enterobacteriaceae Isolates. J. Antimicrob. Chemother. 2011, 66, 48–53. [Google Scholar] [CrossRef]
- Akolkar, N.P.; Adhyapak, J.P.; Aradhye, J.D.; Kumbhani, A.S.; Panchal, B.M.; Jivani, J.K.; Samanta, B.; Pal, R.K.; Thennati, R. Preparation of Amino glycoside Acyl Cyanopyrrolidine Derivatives for Treating or Preventing Diseases Associated with Dipeptidyl Peptidase IV. WO 2009116067 A2. 24 December 2009.
- Ferreira, S.B.; Sodero, A.C.R.; Cardoso, M.F.C.; Lima, E.S.; Kaiser, C.R.; Silva, F.P.; Ferreira, V.F. Synthesis, biological activity, and molecular modeling studies of 1H-1,2,3-triazole derivatives of carbohydrates as α-glucosidases inhibitors. J. Med. Chem. 2010, 53, 2364–2375. [Google Scholar] [CrossRef]
- Ewing, D.F.; Goethals, G.; Mackenzie, G.; Martin, P.; Ronco, G.; Vanbaelinghem, L.; Villa, P. Novel reversed cyclonucleoside analogues with a D-ribofuranose glycone. Carbohydr. Res. 1999, 321, 190–196. [Google Scholar] [CrossRef]
- Lin, H.-S.; Paquette, L.A. A convenient method for determining the concentration of Grignard reagents. Synth. Commun. 1994, 24, 2503–2506. [Google Scholar] [CrossRef]
- Clinical Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 10th ed.; Approved Standard M07-A10; CLSI: Wayne, PA, USA, 2015. [Google Scholar]
- Lubriks, D.; Zogota, R.; Sarpe, V.A.; Matsushita, T.; Sati, G.C.; Haldimann, K.; Gysin, M.; Böttger, E.C.; Vasella, A.; Suna, E.; et al. Synthesis and Antibacterial Activity of Propylamycin Derivatives Functionalized at the 5′- and Other Positions with a View to Overcoming Resistance due to Aminoglycoside Modifying Enzymes. ACS Infect. Dis. 2021, 7, 2413–2424. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).