

State of Knowledge on the Acquisition, Diversity, Interspecies Attribution and Spread of Antimicrobial Resistance between Humans, Animals and the Environment: A Systematic Review
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
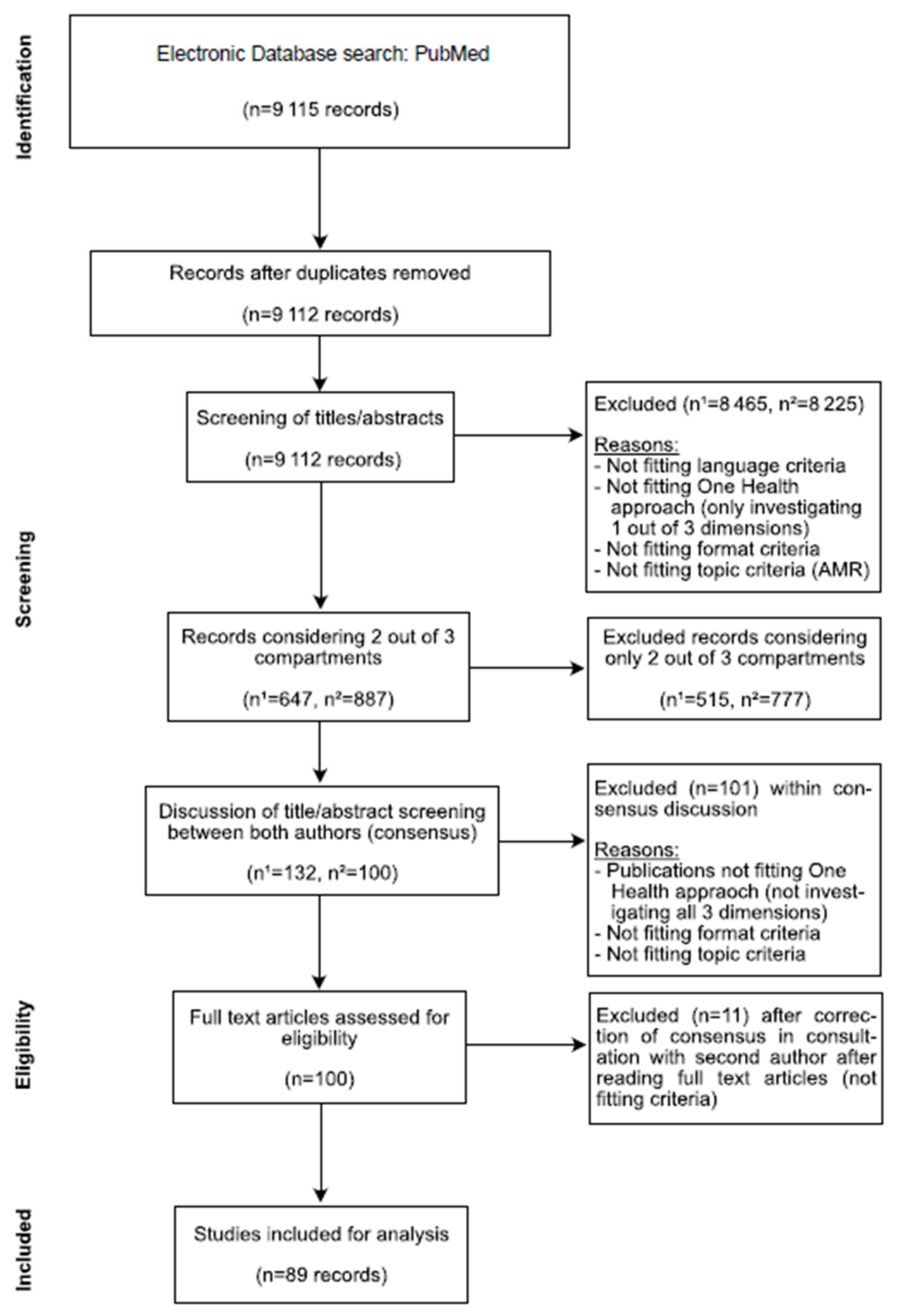
Literature Search
3. Results
3.1. Countries
3.2. Environmental Sectors
3.3. Animal Species
3.4. Human Sector
3.5. Bacteria
3.6. Analysis
3.6.1. Spread of Pheno- and Genotypic Antimicrobial Resistance
3.6.2. Antimicrobial Resistance as a Multifactorial Problem

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor Examined | Key Finding | Comment |
|---|---|---|
| Number of studies | Only about 1 percent of studies consider two or more compartments | Out of 9115 studies |
| Countries | More than 20% of the studies each, were conducted in China and the United States of America | Studies were conducted in 92 countries. |
| Environmental sectors | Most studies were on water (67%), soil (26%), food (20%) and close animal environment (18%) | Other sectors were human hospital environment, close human environment, plant, sludge, sea food and industry |
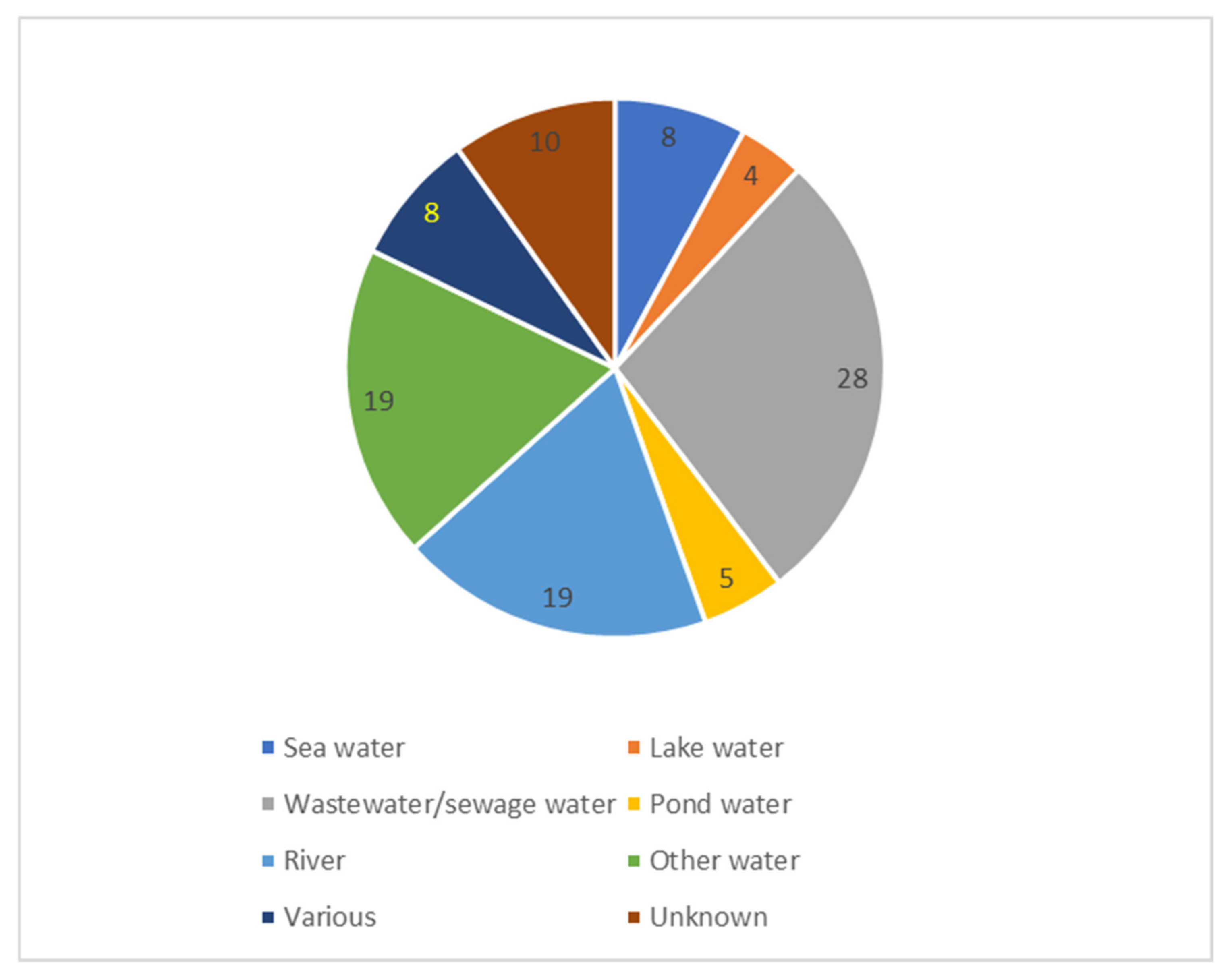
| Water | Wastewater was most often examined (31% of the studies) | Other water sources were sea, lake, pond and river water |
| Animals | Cattle (29% of the studies) and pigs (36%) were the most important individual species | Other animals studied were sheep, goat, dogs, cats, rabbits, insects, rodents, horses, fish and wild birds |
| Meat | Chicken (33%) and pork meat (25%) was most often examined | Other meat was from turkey, dugs, sheep, fish, eggs and milk |
| Human samples | Human samples were mostly clinical samples (65%) | |
| Bacterial species | Salmonella (26%), Escherichia coli (19%) and Enterococci (9%) where the most important identified bacteria | Others were Listeria, Aeromonas, Clostridium, Campylobacter and Pseudomonas. |
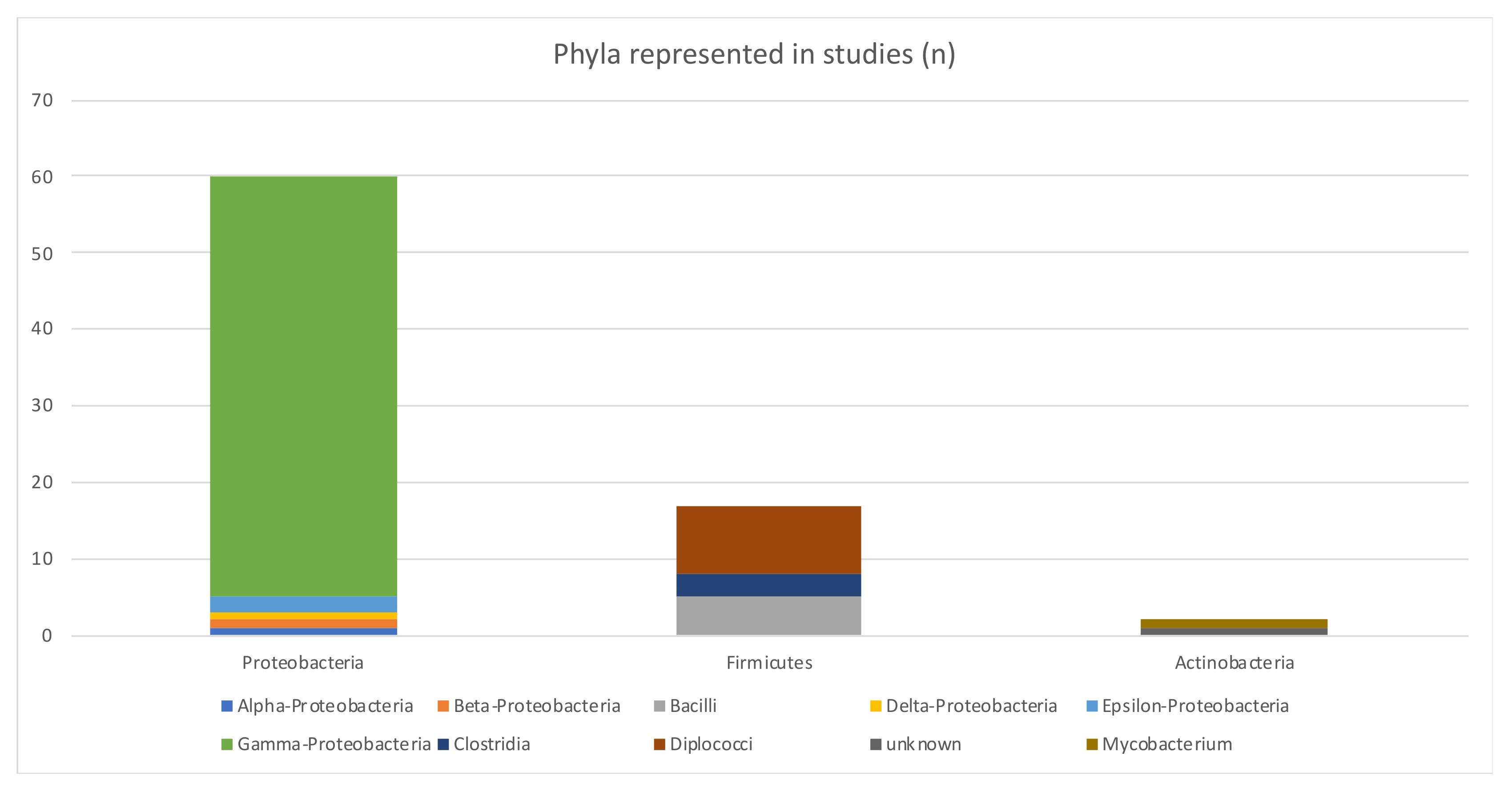
| Bacterial phyla | Proteobacteria (66%) were the most studied phyla | Other phyla were Firmicutes and Actinobacteria |
4. Discussion
Towards Better Integrated Study Design
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Review on Antimicrobial Resistance; Government of the United Kingdom: London, UK, 2016.
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; ColombCotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A populationlevel modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacconelli, E.; Pezzani, M.D. Public health burden of antimicrobial resistance in Europe. Lancet Infect. Dis. 2019, 19, 4–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relman, D.A.; Lipsitch, M. Microbiome as a tool and a target in the effort to address antimicrobial resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 12902–12910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, D.I.; Hughes, D. Selection and Transmission of Antibiotic-Resistant Bacteria. Microbiol. Spectr. 2017, 5, 5.4.12. [Google Scholar] [CrossRef]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014, 509, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yang, X.; Li, J.; Lv, N.; Liu, F.; Wu, J.; Lin, I.Y.; Wu, N.; Weimer, B.C.; Gao, G.F.; et al. The Bacterial Mobile Resistome Transfer Network Connecting the Animal and Human Microbiomes. Appl. Environ. Microbiol. 2016, 82, 6672–6681. [Google Scholar] [CrossRef] [Green Version]
- Smillie, C.S.; Smith, M.B.; Friedman, J.; Cordero, O.X.; David, L.A.; Alm, E.J. Ecology drives a global network of gene exchange connecting the human microbiome. Nature 2011, 480, 241–244. [Google Scholar] [CrossRef]
- Woolhouse, M.; Ward, M.; Van Bunnik, B.; Farrar, J. Antimicrobial resistance in humans, livestock and the wider environment. Philos. Trans. R Soc. Lond. B Biol. Sci. 2015, 370, 1670. [Google Scholar] [CrossRef]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G. The structure and diversity of human, animal and environmental resistomes. Microbiome 2016, 4, 54. [Google Scholar] [CrossRef] [Green Version]
- Aenishaenslin, C.; Haesler, B.; Ravel, A.; Parmley, J.; Stark, K.; Buckeridge, D. Evidence needed for antimicrobial resistance surveillance systems. Bull. World Health Organ. 2019, 97, 283–289. [Google Scholar] [CrossRef]
- Larsson, D.G.J.; Andremont, A.; Bengtsson-Palme, J.; Brandt, K.K.; De Roda Husman, A.M.; Fagerstedt, P.; Fick, J.; Flach, C.F.; Gaze, W.H.; Kuroda, M.; et al. Critical knowledge gaps and research needs related to the environmental dimensions of antibiotic resistance. Environ. Int. 2018, 117, 132–138. [Google Scholar] [CrossRef]
- Rousham, E.K.; Unicomb, L.; Islam, M.A. Human, animal and environmental contributors to antibiotic resistance in low-resource settings: Integrating behavioural, epidemiological and One Health approaches. Proc. Biol. Sci. 2018, 285, 20180332. [Google Scholar] [CrossRef] [Green Version]
- Interagency Coordination Group on Antimicrobial Resistance. Surveillance and Monitoring for Antimicrobial Use and Resistance; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Interagency Coordination Group on Antimicrobial Resistance. No Time to Wait: Securing the Future from Drug-Reistant Infections: Report to the Secretary-General of the United Nations; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Robinson, T.P.; Bu, D.P.; Carrique-Mas, J.; Fèvre, E.M.; Gilbert, M.; Grace, D.; Hay, S.I.; Jiwakanon, J.; Kakkar, M.; Kariuki, S.; et al. Antibiotic resistance is the quintessential One Health issue. Trans. R Soc. Trop. Med. Hygen. 2016, 110, 377–380. [Google Scholar] [CrossRef] [Green Version]
- Zinsstag, J.; Schelling, E.; Waltner-Toews, D.; Tanner, M. From one medicine to one health and systemic approaches to health and well-being. Prev. Vet. Med. 2011, 101, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, E. A diagnostic approach going beyond panaceas. Proc. Natl. Acad. Sci. USA 2007, 104, 15181–15187. [Google Scholar] [CrossRef] [Green Version]
- International Environmental AMR Forum. Initiatives for Addressing Antimicrobial Resistance in the Environment: Current Situation and Challenges; Wellcome Trust, U.S. Centers for Disease Control and Prevention, UK Science & Innovation Network: London, UK, 2018. [Google Scholar]
- Dhaka, P.; Vijay, D.; Vergis, J.; Negi, M.; Kumar, M.; Mohan, V.; Doijad, S.; Poharkar, K.V.; Malik, S.S.; Barbuddhe, S.B.; et al. Genetic diversity and antibiogram profile of diarrhoeagenic Escherichia coli pathotypes isolated from human, animal, foods and associated environmental sources. Infect. Ecol. Epidemiol. 2016, 6, 31055. [Google Scholar] [CrossRef]
- Dulo, F.; Feleke, A.; Szonyi, B.; Fries, R.; Baumann, M.P.; Grace, D. Isolation of Multidrug-Resistant Escherichia coli O157 from Goats in the Somali Region of Ethiopia: A Cross-Sectional, Abattoir-Based Study. PLoS ONE 2015, 10, e0142905. [Google Scholar] [CrossRef]
- Pehrsson, E.C.; Tsukayama, P.; Patel, S.; Mejía-Bautista, M.; Sosa-Soto, G.; Navarrete, K.M.; Calderon, M.; Cabrera, L.; Hoyos-Arango, W.; Bertoli, M.T.; et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature 2016, 533, 212–216. [Google Scholar] [CrossRef]
- Carruth, L.; Roess, A.A.; Terefe, Y.; Hosh, F.M.; Salman, M.D. Antimicrobial resistance and food safety in Africa. Lancet Infect. Dis. 2017, 17, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Busani, L.; Graziani, C.; Battisti, A.; Franco, A.; Ricci, A.; Vio, D.; Digiannatale, E.; Paterlini, F.; D’Incau, M.; Owczarek, S.; et al. Antibiotic resistance in Salmonella enterica serotypes Typhimurium, Enteritidis and Infantis from human infections, foodstuffs and farm animals in Italy. Epidemiol. Infect. 2004, 132, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Atterby, C.; Borjesson, S.; Ny, S.; Jarhult, J.D.; Byfors, S.; Bonnedahl, J. ESBL-producing Escherichia coli in Swedish gulls-A case of environmental pollution from humans? PLoS ONE 2017, 12, e0190380. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.R.; Elghaieb, H.; Leon-Sampedro, R.; Abbassi, M.S.; Novais, C.; Coque, T.M.; Hassen, A.; Peixe, L. Detection of optrA in the African continent (Tunisia) within a mosaic Enterococcus faecalis plasmid from urban wastewaters. J. Antimicrob. Chemother. 2017, 72, 3245–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keelara, S.; Thakur, S. Dissemination of plasmid-encoded AmpC beta-lactamases in antimicrobial resistant Salmonella serotypes originating from humans, pigs and the swine environment. Vet. Microbiol. 2014, 173, 76–83. [Google Scholar] [CrossRef]
- Esteve, C.; Alcaide, E.; Blasco, M.D. Aeromonas hydrophila subsp. dhakensis isolated from feces, water and fish in Mediterranean Spain. Microbes Environ. 2012, 27, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Chan, E.W.; Xie, M.; Ye, L.; Dong, N.; Chen, S. Widespread distribution of mcr-1-bearing bacteria in the ecosystem, 2015 to 2016. Euro Surveill 2017, 22, 17-00206. [Google Scholar] [CrossRef] [Green Version]
- Field, W.; Hershberg, R. Alarmingly High Segregation Frequencies of Quinolone Resistance Alleles within Human and Animal Microbiomes Are Not Explained by Direct Clinical Antibiotic Exposure. Genome Biol. Evol. 2015, 7, 1743–1757. [Google Scholar] [CrossRef] [Green Version]
- Gatica, J.; Jurkevitch, E.; Cytryn, E. Comparative Metagenomics and Network Analyses Provide Novel Insights Into the Scope and Distribution of beta-Lactamase Homologs in the Environment. Front. Microbiol. 2019, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.W.; Thawng, C.N.; Lee, K.; Cha, C.J. Revisiting Polymorphic Diversity of Aminoglycoside N-Acetyltransferase AAC(6′)-Ib Based on Bacterial Genomes of Human, Animal, and Environmental Origins. Front. Microbiol. 2018, 9, 1831. [Google Scholar] [CrossRef]
- Mutters, N.T.; Bieber, C.P.; Hauck, C.; Reiner, G.; Malek, V.; Frank, U. Comparison of livestock-associated and health care-associated MRSA-genes, virulence, and resistance. Diagn. Microbiol. Infect. Dis. 2016, 86, 417–421. [Google Scholar] [CrossRef]
- Kyselkova, M.; Chronakova, A.; Volna, L.; Nemec, J.; Ulmann, V.; Scharfen, J.; Elhottova, D. Tetracycline resistance and presence of tetracycline resistance determinants tet(V) and tap in rapidly growing mycobacteria from agricultural soils and clinical isolates. Microbes Environ. 2012, 27, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.L. Bottlenecks in the transferability of antibiotic resistance from natural ecosystems to human bacterial pathogens. Front. Microbiol. 2012, 2, 265. [Google Scholar] [CrossRef] [Green Version]
- Perry, J.A.; Wright, G.D. The antibiotic resistance mobilome: Searching for the between environment and clinic. Front. Microbiol. 2013, 4, 138. [Google Scholar] [CrossRef] [Green Version]
- D’Costa, V.; McGrann, K.; Hughes, D.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef] [Green Version]
- D’Costa, V.M.; Gri?ths, E.; Wright, G.D. Expanding the soil antibiotic resistome: Exploring environmental diversity. Curr. Opin. Microbiol. 2007, 10, 481–489. [Google Scholar] [CrossRef]
- Kuehn, I.; Iversen, A.; Finn, M.; Greko, C.; Burman, L.G.; Blanch, A.R.; Vilanova, X.; Manero, A.; Taylor, H.; Caplin, J.; et al. Occurrence and relatedness of vancomycin-resistant enterococci in animals, humans, and the environment in different European regions. Appl. Environ. Microbiol. 2005, 71, 5383–5390. [Google Scholar] [CrossRef] [Green Version]
- Nesme, J.; Cecillon, S.; Delmont, T.O.; Monier, J.M.; Vogel, T.M.; Simonet, P. Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr. Biol. 2014, 24, 1096–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novais, C.; Freitas, A.R.; Sousa, J.C.; Baquero, F.; Coque, T.M.; Peixe, L.V. Diversity of Tn1546 and its role in the dissemination of vancomycin-resistant enterococci in Portugal. Antimicrob. Agents Chemother. 2008, 52, 1001–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noll, M.; Kleta, S.; Al Dahouk, S. Antibiotic susceptibility of 259 Listeria monocytogenes strains isolated from food, food-processing plants and human samples in Germany. J. Infect. Public Health 2018, 11, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Antunes, P.; Machado, J.; Peixe, L. Characterization of antimicrobial resistance and class 1 and 2 integrons in Salmonella enterica isolates from different sources in Portugal. J. Antimicrob. Chemother. 2006, 58, 297–304. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, X.; Che, J.; Xiong, Y.; Xu, Y.; Zhang, L.; Lan, R.; Xia, L.; Walsh, T.R.; Xu, J.; et al. Detection and dissemination of the colistin resistance gene, mcr-1, from isolates and faecal samples in China. J. Med. Microbiol. 2017, 66, 119–125. [Google Scholar] [CrossRef]
- Hu, J.; Shi, J.; Chang, H.; Li, D.; Yang, M.; Kamagata, Y. Phenotyping and genotyping of antibiotic-resistant Escherichia coli isolated from a natural river basin. Environ. Sci. Technol. 2008, 42, 3415–3420. [Google Scholar] [CrossRef]
- Ibekwe, A.M.; Murinda, S.E.; Graves, A.K. Genetic diversity and antimicrobial resistance of Escherichia coli from human and animal sources uncovers multiple resistances from human sources. PLoS ONE 2011, 6, e20819. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Xia, Y.; Li, B.; Yang, Y.; Li, L.G.; Tiedje, J.M.; Zhang, T. Metagenomic Assembly Reveals Hosts of Antibiotic Resistance Genes and the Shared Resistome in Pig, Chicken, and Human Feces. Environ. Sci. Technol. 2016, 50, 420–427. [Google Scholar] [CrossRef]
- Ma, Y.; Li, M.; Xu, X.; Fu, Y.; Xiong, Z.; Zhang, L.; Qu, X.; Zhang, H.; Wie, Y.; Zhan, Z.; et al. High-levels of resistance to quinolone and cephalosporin antibiotics in MDR-ACSSuT Salmonella enterica serovar Enteritidis mainly isolated from patients and foods in Shanghai, China. Int. J. Food Microbiol. 2018, 286, 190–196. [Google Scholar] [CrossRef]
- Montealegre, M.C.; Roy, S.; Boni, F.; Hossain, M.I.; Navab-Daneshmand, T.; Caduff, L.; Faruque, A.S.G.; Islam, M.A.; Julian, T.R. Risk Factors for Detection, Survival, and Growth of Antibiotic-Resistant and Pathogenic Escherichia coli in Household Soils in Rural Bangladesh. Appl. Environ. Microbiol. 2018, 84, e01978-18. [Google Scholar] [CrossRef] [Green Version]
- Chao, G.; Zhang, X.; Zhang, X.; Huang, Y.; Xu, L.; Zhou, L.; Yang, W.; Jiang, Y.; Xue, F.; Wu, Y. Phenotypic and genotypic characterization of methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible Staphylococcus aureus (MSSA) from different sources in China. Foodborne Pathog. Dis. 2013, 10, 214–221. [Google Scholar] [CrossRef]
- Gallati, C.; Stephan, R.; Hachler, H.; Malorny, B.; Schroeter, A.; Nuesch-Inderbinen, M. Characterization of Salmonella enterica subsp. enterica serovar 4,[5],12:i:- clones isolated from human and other sources in Switzerland between 2007 and 2011. Foodborne Pathog. Dis. 2013, 10, 549–554. [Google Scholar] [CrossRef]
- Kuang, D.; Zhang, J.; Xu, X.; Shi, W.; Yang, X.; Su, X.; Shi, X.; Meng, J. Increase in Ceftriaxone Resistance and Widespread Extended-Spectrum beta-Lactamases Genes Among Salmonella enterica from Human and Nonhuman Sources. Foodborne Pathog. Dis. 2018, 15, 770–775. [Google Scholar] [CrossRef]
- Dawes, F.E.; Kuzevski, A.; Bettelheim, K.A.; Hornitzky, M.A.; Djordjevic, S.P.; Walker, M.J. Distribution of class 1 integrons with IS26-mediated deletions in their 3′-conserved segments in Escherichia coli of human and animal origin. PLoS ONE 2010, 5, e12754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nógrády, N.; Imre, A.; Kostyak, A.; Toth, A.; Nagy, B. Molecular and pathogenic characterization of Salmonella enterica serovar Bovismorbificans strains of animal, environmental, food, and human origin in Hungary. Foodborne Pathog. Dis. 2010, 7, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Bonke, R.; Wacheck, S.; Stuber, E.; Meyer, C.; Martlbauer, E.; Fredriksson-Ahomaa, M. Antimicrobial susceptibility and distribution of beta-lactamase A (blaA) and beta-lactamase B (blaB) genes in enteropathogenic Yersinia species. Microb. Drug Resist. 2011, 17, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rey, C.; Svenson, S.B.; Bravo, L.; Siitonen, A.; Pasquale, V.; Dumontet, S.; Ciznar, I.; Krovacek, K. Serotypes and anti-microbial susceptibility of Plesiomonas shigelloides isolates from humans, animals and aquatic environments in different countries. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, D.; Walsh, F. Antibiotic resistance genes across a wide variety of metagenomes. FEMS Microbiol. Ecol. 2016, 92, fiv168. [Google Scholar] [CrossRef]
- Collignon, P.J.; McEwen, S.A. One Health-Its Importance in Helping to Better Control Antimicrobial Resistance. Trop. Med. Infect. Dis. 2019, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Frazao, M.R.; Andrade, L.N.; Darini, A.L.C.; Falcao, J.P. Antimicrobial resistance and plasmid replicons in Yersinia enterocolitica strains isolated in Brazil in 30 years. Braz J. Infect. Dis. 2017, 21, 477–480. [Google Scholar] [CrossRef]
- Fernandes, M.R.; Moura, Q.; Sartori, L.; Silva, K.C.; Cunha, M.P.; Esposito, F.; Lopes, R.; Otutumi, L.K.; Gonçalves, D.D.; Dropa, M.; et al. Silent dissemination of colistin-resistant Escherichia coli in South America could contribute to the global spread of the mcr-1 gene. Euro Surveill 2016, 21, 30214. [Google Scholar] [CrossRef]
- Thong, K.L.; Ang, C.P. Genotypic and phenotypic differentiation of Salmonella enterica serovar Paratyphi B in Malaysia. Southeast Asian J. Trop Med. Public Health 2011, 42, 1178–1189. [Google Scholar]
- Davis, J.A.; Jackson, C.R. Comparative antimicrobial susceptibility of Listeria monocytogenes, L. innocua, and L. welshimeri. Microb. Drug Resist. 2009, 15, 27–32. [Google Scholar] [CrossRef]
- Deredjian, A.; Colinon, C.; Brothier, E.; Favre-Bonte, S.; Cournoyer, B.; Nazaret, S. Antibiotic and metal resistance among hospital and outdoor strains of Pseudomonas aeruginosa. Res. Microbiol. 2011, 162, 689–700. [Google Scholar] [CrossRef]
- Gonzalez, M.; Afonso, O.; Tejedor, M.T. Antimicrobial susceptibility and molecular typing of Enterococcus faecium isolated from humans, chickens and environment in Canary Islands (Spain). Rev. Esp. Quim. 2009, 22, 120–126. [Google Scholar]
- Zeng, J.; Pan, Y.; Yang, J.; Hou, M.; Zeng, Z.; Xiong, W. Metagenomic insights into the distribution of antibiotic resistome between the gut-associated environments and the pristine environments. Environ. Int. 2019, 126, 346–354. [Google Scholar] [CrossRef]
- Savic, D.; Miljkovic-Selimovic, B.; Lepsanovic, Z.; Tambur, Z.; Konstantinovic, S.; Stankovic, N.; Ristanovic, E. Antimicrobial susceptibility and beta-lactamase production in Bacillus cereus isolates from stool of patients, food and environment samples. Vojn. Pregl. 2016, 73, 904–909. [Google Scholar] [CrossRef]
- De la Prieta, M.C.; Francia, M.V.; Seoane, A.; Lobo, J.M.G. Characterization of defective beta-lactamase genes in Yersinia enterocolitica. J. Antimicrob. Chemother. 2006, 58, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Gamboa-Coronado Mdel, M.; Mau-Inchaustegui, S.; Rodriguez-Cavallini, E. Molecular characterization and antimicrobial resistance of Clostridium perfringens isolates of different origins from Costa Rica. Rev. Biol. Trop. 2011, 59, 1479–1485. [Google Scholar]
- Lupindu, A.M.; Dalsgaard, A.; Msoffe, P.L.; Ngowi, H.A.; Mtambo, M.M.; Olsen, J.E. Transmission of antibiotic-resistant Escherichia coli between cattle, humans and the environment in peri-urban livestock keeping communities in Morogoro, Tanzania. Prev. Vet. Med. 2015, 118, 477–482. [Google Scholar] [CrossRef]
- Hayford, A.E.; Brown, E.W.; Zhao, S.; Mammel, M.K.; Gangiredla, J.; Abbott, J.W.; Friedman, S.L.; Ayers, S.L.; Lewis, J.L.; Lacher, D.W.; et al. Genetic and resistance phenotypic subtyping of Salmonella Saintpaul isolates from various food sources and humans: Phylogenetic concordance in combinatory analyses. Infect. Genet. Evol. 2015, 36, 92–107. [Google Scholar] [CrossRef]
- Lindmark, H.; Harbom, B.; Thebo, L.; Andersson, L.; Hedin, G.; Osterman, B.; Lindberg, T.; Andersson, Y.; Westoeoe, A.; Olsson Engvall, E. Genetic characterization and antibiotic resistance of Campylobacter jejuni isolated from meats, water, and humans in Sweden. J. Clin. Microbiol. 2004, 42, 700–706. [Google Scholar] [CrossRef] [Green Version]
- On, S.L.; Nielsen, E.; Engberg, J.; Madsen, M. Validity of SmaI-defined genotypes of Campylobacter jejuni examined by SalI, KpnI, and BamHI polymorphisms: Evidence of identical clones infecting humans, poultry, and cattle. Epidemiol. Infect. 1998, 120, 231–237. [Google Scholar] [CrossRef]
- Janam, R.; Gulati, A.K.; Nath, G. Antibiogram and genotyping of Pseudomonas aeruginosa isolated from human, animal, plant, water and soil sources in north India. Southeast Asian J. Trop. Med. Public Health 2011, 42, 1477–1488. [Google Scholar] [PubMed]
- Knetsch, C.W.; Kumar, N.; Forster, S.C.; Connor, T.R.; Browne, H.P.; Harmanus, C.; Sanders, I.M.; Harris, S.R.; Turner, L.; Morris, T.; et al. Zoonotic Transfer of Clostridium difficile Harboring Antimicrobial Resistance between Farm Animals and Humans. J. Clin. Microbiol. 2018, 56, e01384-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escher, N.A.; Muhummed, A.M.; Hattendorf, J.; Vonaesch, P.; Zinsstag, J. Systematic review and meta-analysis of integrated studies on antimicrobial resistance genes in Africa—A One Health perspective. Trop. Med. Int. Health 2021, 26, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Modarai, M.; Naylor, N.R.; Boyd, S.E.; Atun, R.; Barlow, J.; Holmes, A.H.; Johnson, A.; Robotham, J.V. Quantifying drivers of antibiotic resistance in humans: A systematic review. Lancet Infect. Dis. 2018, 18, e368–e378. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meier, H.; Spinner, K.; Crump, L.; Kuenzli, E.; Schuepbach, G.; Zinsstag, J. State of Knowledge on the Acquisition, Diversity, Interspecies Attribution and Spread of Antimicrobial Resistance between Humans, Animals and the Environment: A Systematic Review. Antibiotics 2023, 12, 73. https://doi.org/10.3390/antibiotics12010073
Meier H, Spinner K, Crump L, Kuenzli E, Schuepbach G, Zinsstag J. State of Knowledge on the Acquisition, Diversity, Interspecies Attribution and Spread of Antimicrobial Resistance between Humans, Animals and the Environment: A Systematic Review. Antibiotics. 2023; 12(1):73. https://doi.org/10.3390/antibiotics12010073
Chicago/Turabian StyleMeier, Hélène, Keira Spinner, Lisa Crump, Esther Kuenzli, Gertraud Schuepbach, and Jakob Zinsstag. 2023. "State of Knowledge on the Acquisition, Diversity, Interspecies Attribution and Spread of Antimicrobial Resistance between Humans, Animals and the Environment: A Systematic Review" Antibiotics 12, no. 1: 73. https://doi.org/10.3390/antibiotics12010073
APA StyleMeier, H., Spinner, K., Crump, L., Kuenzli, E., Schuepbach, G., & Zinsstag, J. (2023). State of Knowledge on the Acquisition, Diversity, Interspecies Attribution and Spread of Antimicrobial Resistance between Humans, Animals and the Environment: A Systematic Review. Antibiotics, 12(1), 73. https://doi.org/10.3390/antibiotics12010073