Abstract
The gastrointestinal tracts of dairy calves and cows are reservoirs of antimicrobial-resistant bacteria (ARB), which are present regardless of previous antimicrobial therapy. Young calves harbor a greater abundance of resistant bacteria than older cows, but the factors driving this high abundance are unknown. Here, we aimed to fully characterize the genomes of multidrug-resistant (MDR) and antimicrobial-susceptible Escherichia coli strains isolated from pre-weaned calves, post-weaned calves, dry cows, and lactating cows and to identify the accessory genes that are associated with the MDR genotype to discover genetic targets that can be exploited to mitigate antimicrobial resistance in dairy farms. Results indicated that both susceptible and resistant E. coli isolates recovered from animals on commercial dairy operations were highly diverse and encoded a large pool of virulence factors. In total, 838 transferrable antimicrobial resistance genes (ARGs) were detected, with genes conferring resistance to aminoglycosides being the most common. Multiple sequence types (STs) associated with mild to severe human gastrointestinal and extraintestinal infections were identified. A Fisher’s Exact Test identified 619 genes (ARGs and non-ARGs) that were significantly enriched in MDR isolates and 147 genes that were significantly enriched in susceptible isolates. Significantly enriched genes in MDR isolates included the iron scavenging aerobactin synthesis and receptor genes (iucABCD-iutA) and the sitABCD system, as well as the P fimbriae pap genes, myo-inositol catabolism (iolABCDEG-iatA), and ascorbate transport genes (ulaABC). The results of this study demonstrate a highly diverse population of E. coli in commercial dairy operations, some of which encode virulence genes responsible for severe human infections and resistance to antibiotics of human health significance. Further, the enriched accessory genes in MDR isolates (aerobactin, sit, P fimbriae, and myo-inositol catabolism and ascorbate transport genes) represent potential targets for reducing colonization of antimicrobial-resistant bacteria in the calf gut.
1. Introduction
Multidrug-resistant (MDR) Escherichia coli are frequently isolated from dairy cow and calf feces, unpasteurized cow’s milk, and culled dairy cow beef and are a major component of the suite of antimicrobial-resistant bacteria (ARB) carried by young calves [1,2,3]. A minority of these E. coli are enterohemorrhagic (EHEC) Shiga toxin-encoding strains that can cause moderate to severe gastrointestinal disease, septicemia, hemolytic uremia syndrome (HUS), chronic sequelae, and possibly death [4,5]. However, E. coli is highly diverse, with many suites of virulence and fitness factors (VFFs) that can cause both mild to severe gastrointestinal and extra-intestinal infections, such as urinary tract infections (UTIs) and meningitis [6]. Major non-EHEC strains causing human disease are typically classified into pathovars (pathotypes) based on the presence of different suites of virulence factors; strains that contain overlapping pathovar suites of these genes are considered hybrid pathovars. Dairy animals are known reservoirs of these MDR or antimicrobial-susceptible pathogens, but most studies of E. coli from these animals have either targeted only a few farms, have focused on a specific resistance phenotype/profile or pathovar, or have not utilized a genome-wide scale approach to evaluate the diversity and dynamics of these organisms in the dairy farm environment.
Dairy cow and calf feces are known reservoirs of ARGs that can contaminate milk, meat, other animals, animal handlers, and the environment [7,8]. Most studies using sensitive molecular techniques have shown that most, if not all, dairy animals carry antimicrobial-resistant bacteria (ARB) in their lower gastrointestinal system and feces, and this is consistent even in the absence of antimicrobial therapy [9,10,11,12]. Multiple studies have demonstrated that antimicrobial administration results in a transient or no increase in antimicrobial resistance (AMR) in the feces and that animals that were never exposed to these drugs may, at times, have the same level of resistance as conventionally raised animals [13,14,15]. Further, it has been repeatedly demonstrated that young calves typically carry a higher level of resistance than older lactating or dry cows, and this, too, is true in the absence of antimicrobial administration [1,2,8,16,17]. However, these calves are exposed to their dam’s microbial communities during and immediately after birth, as well as the surrounding birthing pen environment, all of which have a lower ratio of resistant to susceptible bacteria than that found in calf feces. The dam, the farm environment, colostrum, and milk/milk replacer are the sources of these ARBs, but not much is known about the genetic factors of these bacteria that influence their enrichment and persistence in the calf gut. This information could be used to potentially identify intervention targets to reduce the carriage of ARBs in dairy animals and possibly other food animals. Here, we used a genome-wide approach to evaluate the AMR profiles and virulence factor diversity of E. coli collected from pre-weaned calves, post-weaned calves, dry cows, and lactating cows on 80 commercial dairy farms. We also identify non-resistance conferring genes that co-occur with the MDR phenotype with the objective of identifying potential intervention targets to reduce the occurrence of MDR E. coli in dairy calves.
2. Materials and Methods
E. coli isolates were selected from a previously published study of antimicrobial resistance in bacteria isolated from the feces of animals in different age groups on 80 commercial dairy farms [1]. Selection was based on farm, animal group, and antibiotic sensitivity. Isolates were provisionally prescreened for antibiotic sensitivity by patch-plating onto eight petri dishes, each supplemented with a different class of antibiotics. Based on these data, one isolate per resistance group (MDR or susceptible), per animal group (pre-weaned calves, post-weaned calves, lactating cows, and dry cows), and per farm was identified. This resulted in eight groups of isolates (susceptible dry cow isolates = 315; MDR dry cow isolates = 52; susceptible lactating cow isolates = 1033; MDR lactating cow isolates = 136; susceptible post-weaned calf isolates = 221; MDR post-weaned calf isolates = 140; susceptible pre-weaned calf isolates = 132; MDR pre-weaned calf isolates = 284). Within each of these groups, random numbers were assigned to each isolate using a non-redundant random number generator in Microsoft Excel v. 16.77.1 (Microsoft Corporation, Redmond, WA, USA), and these numbers were reordered from smallest to largest, and the smallest number for each animal group within each farm was selected for genome sequencing.
Selected isolates were grown overnight at 37 °C in L broth (per 1 L: enzymatic digest of casein 10 g, yeast extract 5 g, sodium chloride 5 g), and DNA was extracted from these overnight cultures using a QiaCube platform (Qiagen, Hilden, Germany). Genome sequencing libraries were constructed using a Nextera XT kit (Illumina, La Jolla, CA, USA), and 2 × 150 bp paired-end sequencing was conducted on a NextSeq 500 platform (Illumina) with a High Output flow cell. After demultiplexing the data, reads were trimmed of adaptors, sequencing contaminants, and phiX sequences using DeconSeq [18] and then trimmed for quality and length using Trimmomatic (LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:36) [19]. These cleaned and curated reads were assembled using SPAdes V. 3.14.1 [20]. The genome sequencing data have been deposited at NCBI (Supplementary File S4).
Core genome SNPs were identified by aligning the 264 E. coli genomes used in this study with 118 publicly available Escherichia genomes representing the major phylogenetic groups (A, B1, B2, C, D, E, F, and G) and the near neighbors, E. fergussoni and E. albertii, downloaded from NCBI using the Harvest package [21]. ParSNP was run with the parameters -c and -x and the complete chromosome of E. coli K-12 substrate MG1655 (NCBI accession: NC_000913.3) as the reference genome (-r). Identified SNPs were used to infer a maximum likelihood tree with 1000 bootstrap replicates under default settings using RAxML [22].
Genome sequences were interrogated for transferrable ARGs, as well as SNPs, conferring resistance to antibiotics using the ResFinder 4.1 database with default settings (threshold for %ID = 90%, minimum length = 60%) [23]. Since our aim was to focus on ARGs, an isolate that was provisionally categorized as phenotypically susceptible but was found to carry an ARG (or ARGs associated with resistance to less than three classes of antimicrobials) was reclassified as “resistant” (R) and replaced with a phenotypically susceptible isolate from the same animal group and farm. This substituted isolate was then genomically confirmed as ARG-free. The same approach was taken to genomically validate the presence of ARGs and SNPs conferring resistance to three or more classes of antibiotics in provisionally phenotypical MDR isolates. A genome that encoded ARGs or SNPs conferring resistance to one or two classes of antibiotics was classified as “resistant” (R) and was included in that group for some downstream analyses. The final number of genome sequences used for this study was 108 multidrug-resistant (MDR), 36 antimicrobial-resistant (R), and 120 antimicrobial-susceptible (S) sequences.
Genotypic MDR genomes were identified as those genomes that encoded genes and SNPs known to confer resistance to at least three classes of antibiotics (identified as “MDR” in the analysis). Genotypic antibiotic-resistant isolates were those encoding genes or SNPs known to confer resistance to one or two classes of antibiotics (identified as “R”). Genotypic susceptible genomes were those that did not encode any known genes or SNPs that confer resistance to any antibiotics (identified as “S”). Isolates were not initially selected for sequencing based on resistance profile but were selected based on whether they were provisionally resistant to three or more antibiotics or no antibiotics, and, thus, the ARG content reflects an unbiased selection of MDR isolates. Sequence types (STs) and plasmid replicons were identified using MLST 2.0 [24,25] and PlasmidFinder 2.1 [26] with default settings. For genomes that did not match an exact ST in the MLST 2.0, the raw reads were uploaded to the Enterobase database for novel ST assignment [27].
Since virulence genes are numerous, highly diverse, and do not exclusively function to cause an infection within a mammalian host but also at times aid in survival within and outside of the host, we labeled these genes and the known major virulence factors collectively as “virulence and fitness factors” (VFF). VFFs were identified with ABRicate under default settings [28].
Identification of the pangenome was conducted by annotating the assembled genomes in PROKKA [29] under default settings and then uploading the gff3 files into Roary [30] under default settings. Using this pangenome, genes that were enriched (significantly more abundant) in MDR, as well as susceptible genomes, were identified using a Fisher’s Exact Test with the package “exact2 × 2” in R. To limit the proportion of falsely significant results, q-values (FDR—False Discovery Rate) [31] were estimated for all genes using the package “qvalue” in R (q-values are the proportion of genes that are identified as significant that are estimated to be falsely significant). A q-value threshold of <0.05 was used to identify significant features. Signal proteins were identified among the translated enriched genes to identify secreted proteins using SignalP 5.0 [32].
To visualize the distances between the accessory gene content (present in <99% of genomes) of MDR and susceptible genomes, a non-metric multidimensional scaling (NMDS) analysis using the Jaccard distance metric was inferred, followed by an analysis of similarities (ANOSIM) of the pangenomes of these two groups with and without ARGs included in the analyses using the R package “vegan”. To determine if MDR or S genomes were randomly distributed among the phylogenetic groups, a χ2 test was conducted in R.
3. Results
3.1. Antimicrobial Resistance Genes
In total, there were 838 ARGs detected, 46 of which were unique genes. Genomes encoded genes conferring resistance to between 0 and 8 classes of antibiotics (Table 1). The median and average number of classes to which MDR isolates were resistant were 5 and 4.6, respectively. Isolate genomes encoded between 0 and 16 ARGs, including resistance-conferring SNPs. Among the MDR isolates, the median and average numbers of ARGs per isolate were 7 and 7.3, respectively. In total, 42 resistance-conferring SNPs were detected in a total of 31 genomes. Among the genomes in which these SNPs were detected, the maximum number of SNPs detected was five and the median was 1.

Table 1.
Isolation source, phylogenetic group, multi-locus sequence type (MLST), antimicrobial resistance genes (ARGs) separated by antimicrobial class, antimicrobial resistance-conferring point mutations, metal resistance genes (MRGs), and biocide resistance genes (BRGs) identified among the study isolates. Ami = aminoglycosides, β-Lac = β-Lactams, Flu = fluoroquinolones, Fos = Fosfomycin, MLS = macrolide-lincosamide-streptogramin B, Phe = phenicols, Sul = sulfonamide, Tet = tetracyclines, Tri = trimethoprim. As = arsenic, Cu = copper, Hg = mercury, Ag = silver, Te = Tellurium. BRGs = Biocide resistance genes. QAC = quaternary ammonium compounds.
There was a total of 341 aminoglycoside, 140 tetracycline, 134 sulfonamide, 115 β-lactam, 50 phenicol, 10 macrolide-lincosamide-streptogramin B (MLS), five fosfomycin, and one fluoroquinolone resistance genes detected in all the MDR and R genomes (Table 1). Colistin, fusidic acid, glycopeptide, nitroimidazole, oxazolidinone, and rifampicin resistance genes were not detected in any of the genomes. In total, there were 109 isolates that encoded aminoglycoside ARGs, and 101 of these encoded more than one aminoglycoside ARG. There were only 32, 27, and 18 isolates that encoded more than one sulfonamide, β-lactam, or tetracycline ARG, respectively. The ten most frequently detected ARGs, in order of decreasing frequency, were aph(6)-Id (94 isolates), aph(3″)-Ib (93 isolates), sul2 (90 isolates), tetB (66 isolates), blaCMY-2 (65 isolates), aph(3′)-Ia (61 isolates), tetA (58 isolates), sul1 (40 isolates), floR (39 isolates), blaTEM-1B (34), and aadA1 (22 isolates). Multiple transferrable ARGs conferring resistance or reduced susceptibility to antibiotics critical to human health were identified among the MDR isolates. These include the ESBLs blaCMY-2, blaCTX-M-1, blaCTX-M-14, blaCTX-M-15, blaTEM-214, macrolide resistance genes mphA, mphB, and the fluoroquinolone resistance gene aac(6′)-Ib-cr.
Point mutations in housekeeping genes that conferred resistance to antibiotics were also identified among the isolates (Table 1). Specifically, two mutation types were observed in gyrA (gyrA p.S83L and gyrA p.D87N), four in parC (parC p.A56T, parC p.S57T, parC p.S80I, parC p.E84K), one in parE (parE p.S458A), and two in the ampC promoter (ampC promoter n.-32T>A and n.-42C>T). Non-synonymous mutations in gyrA, parC, and parE are known to confer resistance to fluoroquinolones. Of these, the two most abundant were gyrA p.S83L and parC p.A56T, and they were identified in six isolates each. In total, 18 genomes encoded SNPs in the ampC promoter, and none of these encoded SNPs in gyrA, parC, or parE. Six isolates encoded the gyrA p.S83L mutation, and three of these encoded the gyrA p.D87N mutation. Four of the six isolates with a gyrA mutation also encoded mutations in parC, and one of these encoded mutations in parE. Eleven isolates encoded parC mutations, with six of them having parC p.A56T, four having parC p.S80I, three having parC p.S57T, and one having parC p.E84K. The latter isolate simultaneously encoded parC p.A56T and parC p.S80I. One isolate encoded parE p.S458A, and this isolate also encoded gyrA p.S83L, gyrA p.D87N, and parC p.S80I. Of the 31 isolates encoding these SNPs, 29 were the MDR genotype, one encoded one other ARG, and one encoded no other ARGs.
3.2. Biocide and Metal Resistance Genes
Transferrable biocide resistance genes (BRGs) and metal resistance genes (MRGs) were identified in many genomes; MDR genomes typically encoded more of these resistance genes than R or S genomes (Table 1). For each resistance category (MDR, R, and S), at least two genomes encoded copper resistance genes (pco), with five MDR genomes encoding this operon. Ten genomes encoded silver resistance genes (sil). The majority of the mercury resistance-encoding genomes were MDR (43 genomes) compared with R and S genomes (1 genome each). Similarly, the biocide resistance gene, qacEΔ, was found in 42 MDR genomes and 2 R genomes, but it was not found in any S genomes.
3.3. Co-Occurrence between ARGs, MRGs, and BRGs
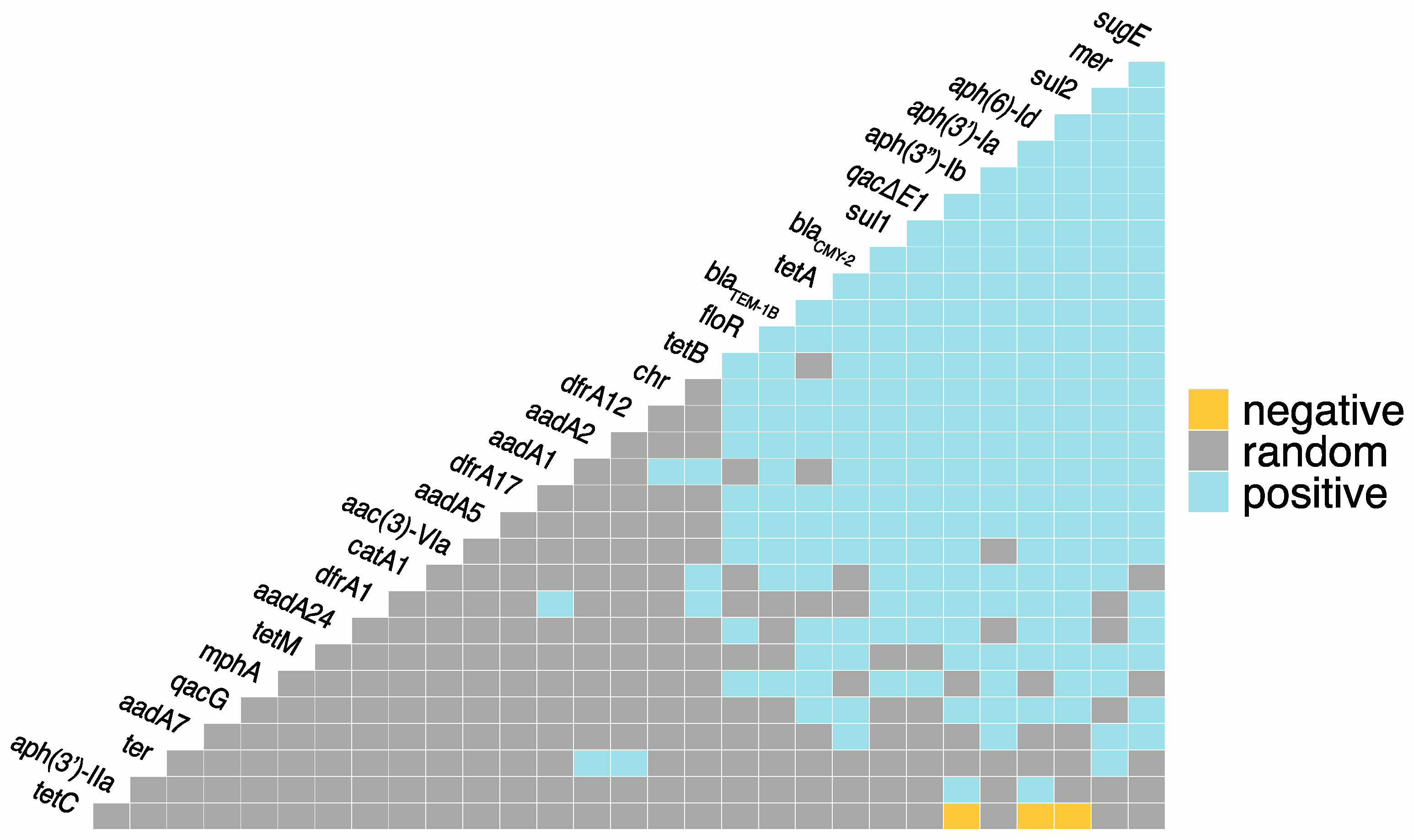
In total, there were 222 positive co-occurrences between and among the identified ARGs, BRGs, and MRGs, and only three negative co-occurrences (Figure 1; Supplementary File S1). The most frequent co-occurrences between ARGs were aph(3″)-Ib-aph(6′)-Id, aph(3″)-Ib-sul2, and aph(6′)-Id-sul2. Azithromycin resistance gene, mphA, had positive co-occurrences with ARGs sul1, sul2, tetA, blaTEM-1B, and floR, as well as BRG qacE∆1 and MRG mer. blaCMY-2 had positive co-occurrences with 18 other ARGs, as well as BRGs qacE∆1, qacG, sugE, and MRGs mer and chr. The most frequent co-occurrences between BRGs were sugE-qacE∆1 and sugE-qacE∆1. Both qacE∆1 and sugE had twenty positive co-occurrences with ARGs. qacE∆1 had the most co-occurrences with sul1, and sugE had the most co-occurrences with blaCMY-2. The only co-occurrences between individual MRGs were mer-ter and mer-chr. Mercury resistance genes (mer) had 20 positive co-occurrences with ARGs as well as sugE and ter. Chromate resistance genes (chr) had positive co-occurrence with 10 ARGs, and tellurium resistance (ter) had positive co-occurrences with two ARGs, aadA2 and dfrA12. The only observed negative co-occurrences were between tetC and aph(3″)-Ib, aph(6′)-Id, and sul2.
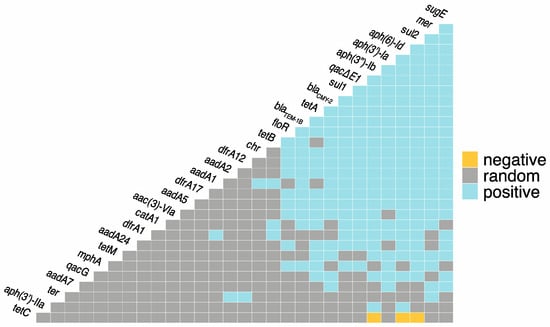
Figure 1.
Co-occurrence among antimicrobial resistance genes (ARGs), metal resistance genes (MRGs), and biocide resistance genes (BRGs). Blue = statistically significant positive co-occurrence between genes. Yellow = statistically significant negative co-occurrence between genes. Grey = random co-occurrence.
3.4. Plasmid Replicon Diversity
There were 708 total plasmid replicons comprising 42 different replicon types identified among the isolates (Table 2). The most frequently detected replicon was IncFIB(AP001918), which was identified in 170 isolates, followed by ColRNAI, IncFIA, IncI1_Alpha, IncFIC(FII), and IncA/C2, which were detected in 92, 87, 47, 37, and 32 genomes, respectively. For all resistance groups (MDR, R, and S), the IncFIB(AP001918) plasmid was the most frequently detected. In terms of the percentage of replicon presence, the greatest difference between MDR and S genomes was in IncFIB(AP001918), which was carried by 65% of S genomes and 40% of MDR genomes. Similarly, appreciable differences were identified in the carriage rates of IncFIC(FII), IncA/C2, and IncFIA between MDR and S genomes (19.1%, 15.8%, and 11.9% difference between MDR and S genomes).
Table 2.
Isolation source, phylogenetic group, sequence type (MLST), resistance group (MDR = multidrug-resistant; R = antimicrobial-resistant; S = antimicrobial-susceptible), plasmid replicons identified in study genomes.
3.5. Genomic Diversity
In total, 142 STs were identified among the isolates, with ST10, ST58, ST88, and ST56 being the most frequently detected, which were identified 20, 18, 10, and 7 times, respectively (Table 1). Among the MDR isolates, ST10 was the most frequently detected ST (13 isolates). Among the susceptible isolates, ST58 was the most frequently detected ST (10 isolates). Among the isolates that were resistant, except for MDR, ST95 was the most frequently detected, followed by ST10 (three and two isolates, respectively). Within the MDR isolates, ST10 was the most frequently detected ST from pre- and post-weaned calves (ST56 was detected as many times as ST10 in post-weaned calves). ST88 was the most frequently detected ST among MDR isolates from lactating cows (five isolates). ST58 was the most frequently detected ST among MDR isolates from dry cows. Within the susceptible isolates, ST58 was the most frequently detected among lactating cow isolates, while ST164 was the most frequently detected among dry cow isolates; ST32 was the most frequently detected among pre-weaned calf isolates, and ST329 and ST2521 were the most frequently detected among post-weaned calf isolates.
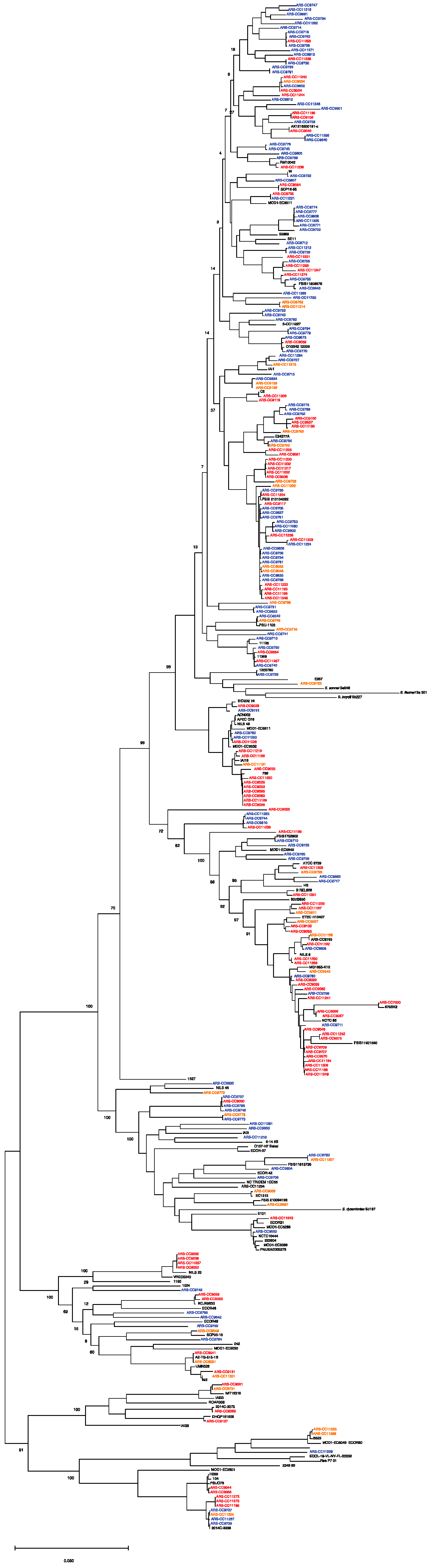
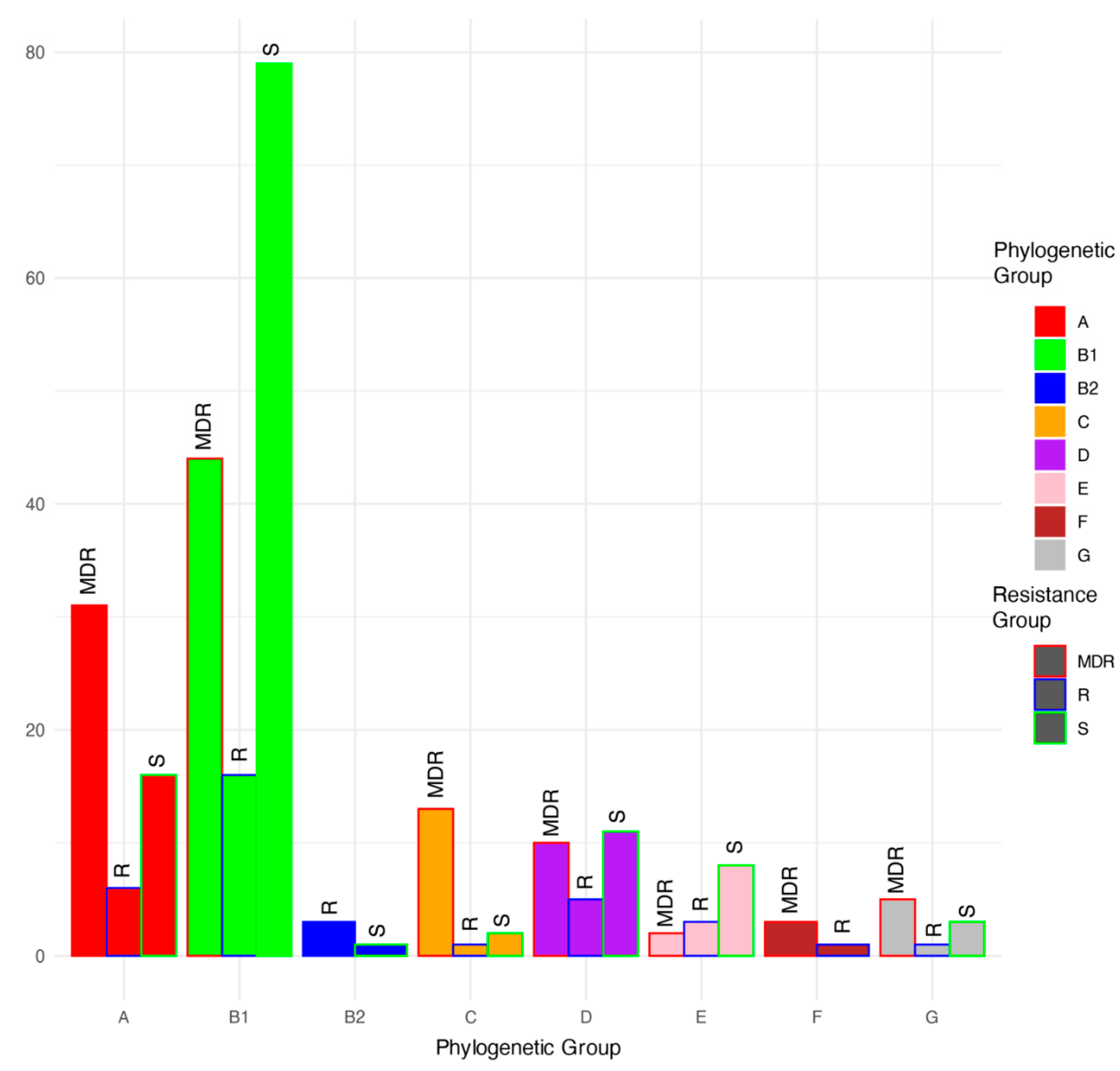
Based on a phylogenetic comparison of the genomes with Escherichia genomes downloaded from NCBI, the isolates were classified into phylogroups A (53 isolates), B1 (139 isolates), B2 (4 isolates), C (16 isolates), D (26 isolates), E (13 isolates), F (4 isolates), and G (9 isolates) (Figure 2 and Figure 3; Table 1). Among the MDR group, isolates were classified as phylogroups B1, A, C, D, G, F, and E and were detected 44, 31, 13, 10, 5, 3, and 2 times, respectively. Among the resistant isolates, B1, A, D, E, B2, C, F, and G were detected 16, 6, 5, 3, 3, 1, 1, and 1 times, respectively. Among the susceptible isolates, B1, A, D, E, G, C, and B2 were detected 79, 16, 11, 8, 3, 2, and 1 times, respectively. MDR genomes were more likely to be phylogenetic groups A and C (residuals = 2.0 and 2.5, respectively), and S genomes were more likely to be phylogenetic group B1 (residual = 1.99; Chi-square test, χ2 = 46.629, p < 0.001).
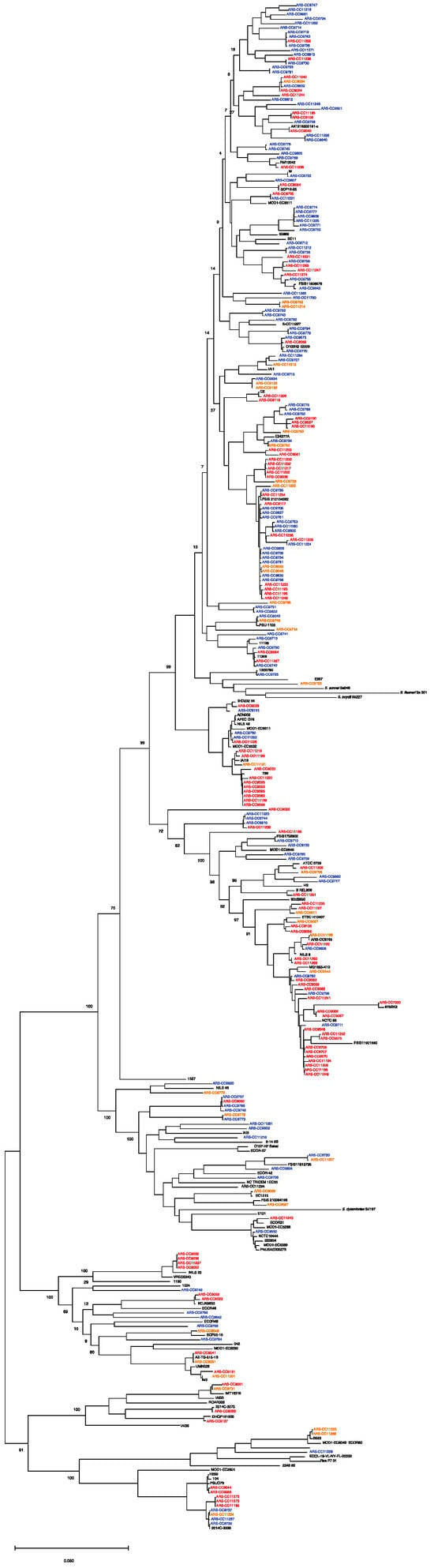
Figure 2.
Maximum likelihood core genome phylogeny of study genomes (n = 264) and reference genomes (n = 94) of the major phylogenetic groups. The tree is rooted in E. albertii. Red = multidrug-resistant genomes. Orange = antimicrobial-resistant genomes. Blue = antimicrobial-susceptible genomes. The maximum likelihood tree was inferred using RaxML with 1000 bootstrap replicates. Bar represents number of substitutions per site.
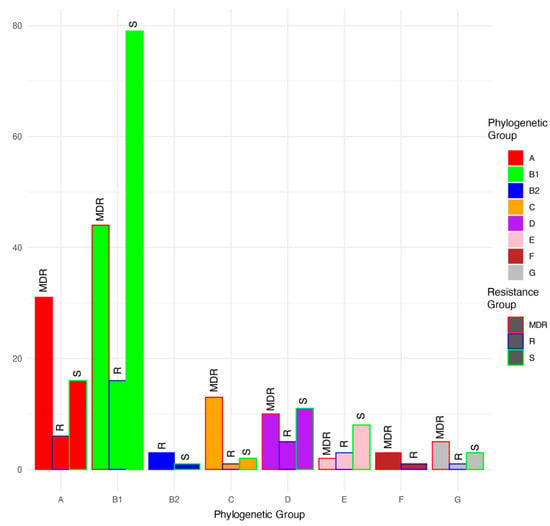
Figure 3.
Proportions (%) of multidrug-resistant (MDR), antimicrobial-resistant (R), and antimicrobial-susceptible (S) isolates within each phylogenetic group.
3.6. Virulence and Fitness Factors
Genomes were interrogated for a broad suite of virulence and fitness factors (VFFs) that have both major and minor roles in pathogenesis, intra-host survival, and between-host transmission. In total, 49,407 VFFs were identified among the isolates, with multiple VFFs identified in all genomes (Supplementary File S2). The mean and median number of VFFs per isolate were 187 and 186, respectively. The highest number of VFFs identified in any isolate was 279, and the lowest was 114.
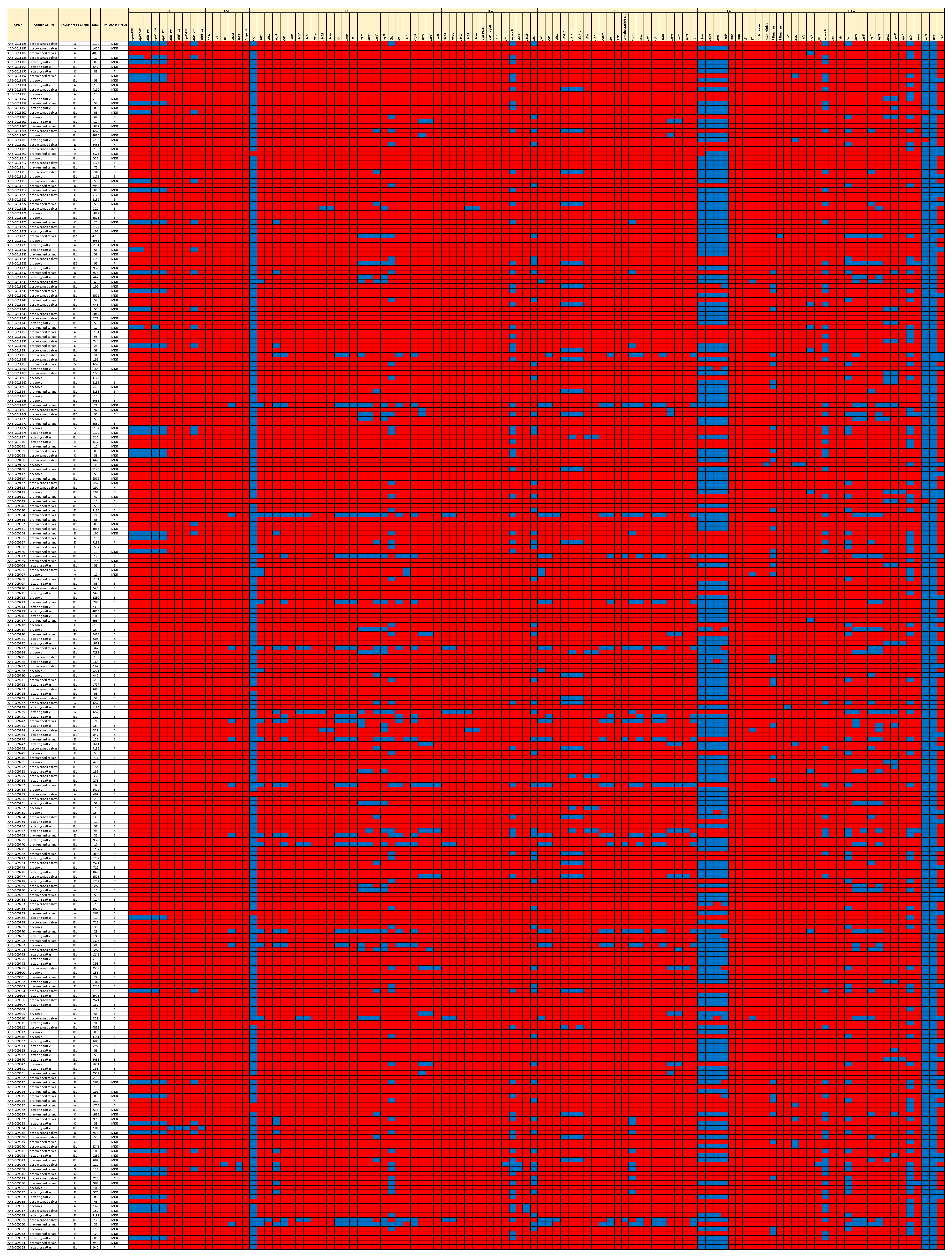
Multiple isolates with VFFs known to play significant roles in the pathogenesis of the major pathovars were detected (Figure 4). The sequences of nine strains (3.4% of the total) significantly aligned with the Shiga toxin genes of Shiga toxigenic E. coli (STEC). Six of these nine genomes encoded stx1A and stx1B, one encoded stx2A, three encoded stx2B, two encoded stx2d, and a single genome encoded stx1A, stx1B, stx2A, and stx2B. Among the STEC genomes, four encoded the intestinal adherence factor, intimin (eae), which is integral in STEC pathogenesis, indicating that these isolates were potential enterohemorrhagic E. coli (EHEC). Two of these, O103:H2 and O45:H2, are considered adulterants by the USDA Food Safety and Inspection Service [33]. Based on the presence of eae in their genomes, 16 isolates were determined to be enteropathogenic E. coli (EPEC). Of these, two were genotypically resistant, five were MDR, and nine were susceptible. Two MDR genomes and two susceptible genomes encoded the heat-stable enterotoxin estIa gene (STa) of enterotoxigenic E. coli (ETEC), but these did not encode the LT heat-labile toxin. Four isolates were characterized as uropathogenic E. coli (UPEC) based on the presence of chuA (heme-binding protein), fyuA (yersiniabactin receptor), and vat (autotransporter serine protease toxin). Two of these were MDR ST117, and two were resistant ST95 [34]. In total, 41 isolates (15% of the total) were identified as extraintestinal pathogenic E. coli (ExPEC) based on the presence of at least two of the following: VFs pap (P fimbriae), sfa/foc (S and F1C fimbriae), afa/draBC (Dr binding adhesins), kpsM (group 2 capsule), and iutA (aerobactin receptor) in their genomes. Thirty-four of these isolates were MDR; three were resistant, and four were susceptible. Among these ExPEC isolates, 16 different STs were identified, with ST10, ST88, and ST973 being the most frequently detected. Multiple other ExPEC genes were detected, such as iroN (salmochelin siderophore receptor), ibeABC (invasin of brain endothelial cells), sitABC (iron transport protein), cnf (cytotoxic necrotizing factor), cdt (cytolethal distending toxin), and pic (serine protease autotransporter) (Figure 4, Supplementary File S2). In total, 60 isolates (22%) were members of STs that are among the most common causes of ExPEC infections, including ST10, ST23, ST38, ST58, ST69, ST88, ST95, ST117, and ST167.

Figure 4.
Presence/absence heatmap of virulence and fitness factors (VFFs) of the major pathovars. Blue = present. Red = absent. MLST = Multi-Locus Sequence Type.
3.7. Accessory Genes Associated with the MDR and Susceptible Genotypes
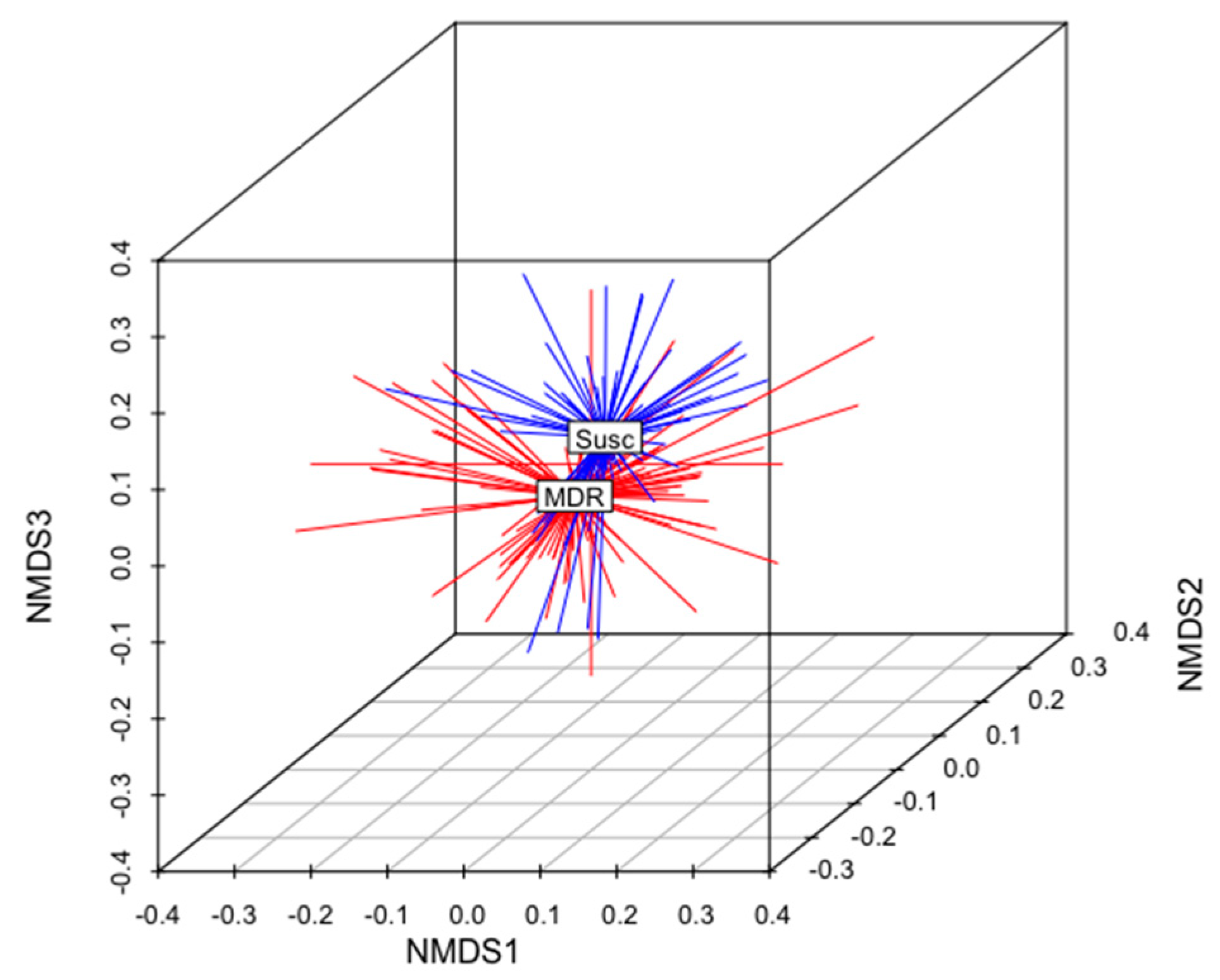
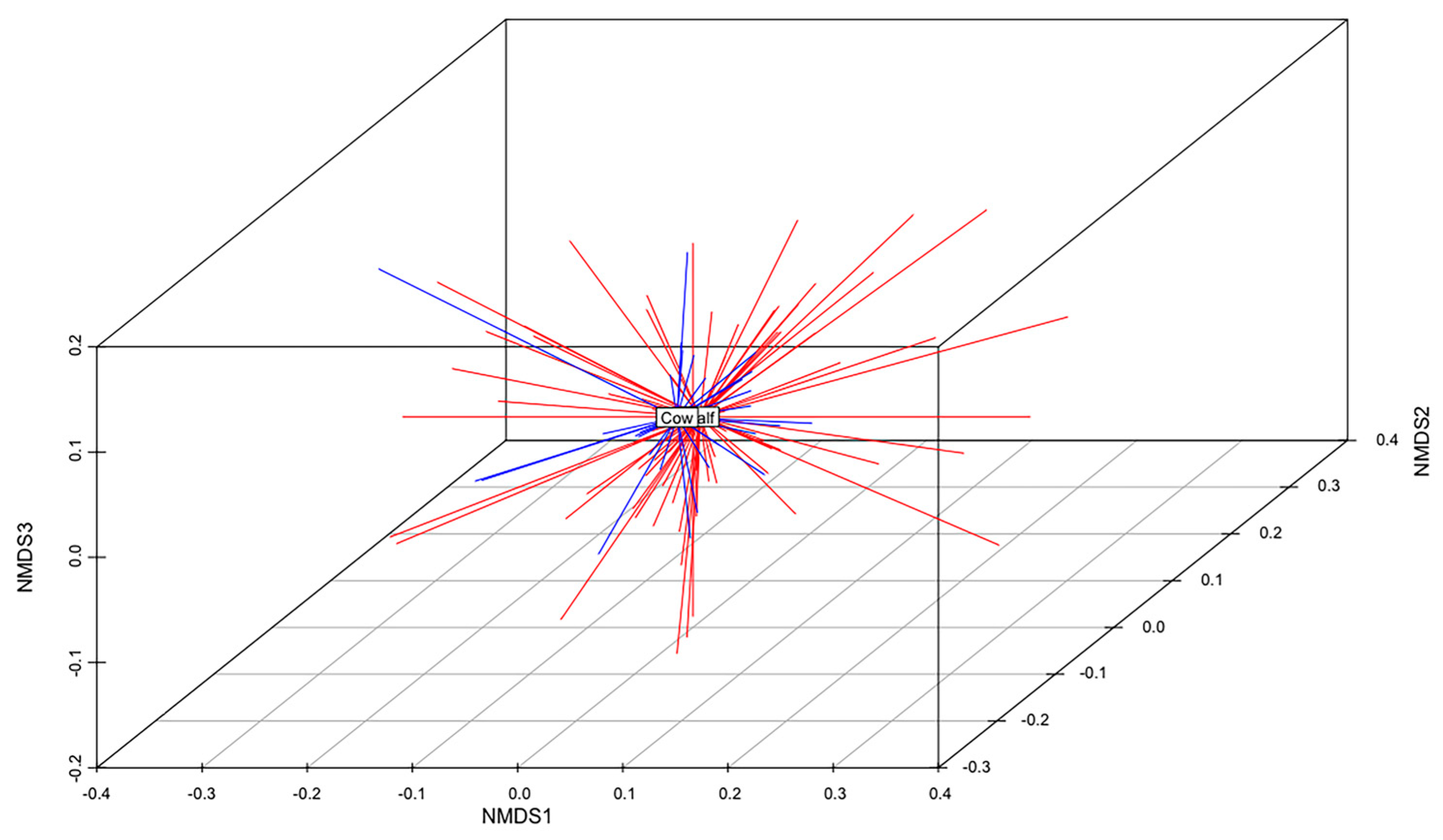
The total gene content of the MDR and S genomes were compared to identify differences in accessory genes between these two groups of isolates. A non-metric multidimensional scaling (NMDS) analysis and an analysis of similarities (anosim) test demonstrated that the gene content of MDR genomes was different than that of S genomes (Figure 5). When all ARGs were removed from this analysis, this relationship remained statistically significant (anosim R = 0.16, p < 0.001), indicating that MDR genomes and susceptible genomes were slightly, yet significantly, different from each other, and these differences were not due to the presence of ARGs (Figure 5). There was no difference in the gene content of MDR E. coli isolated from calves than those isolated from adult cows (anosim p = 0.972), indicating that the gene contents of MDR strains shed by cows were not significantly different than those shed by calves (Figure 6).
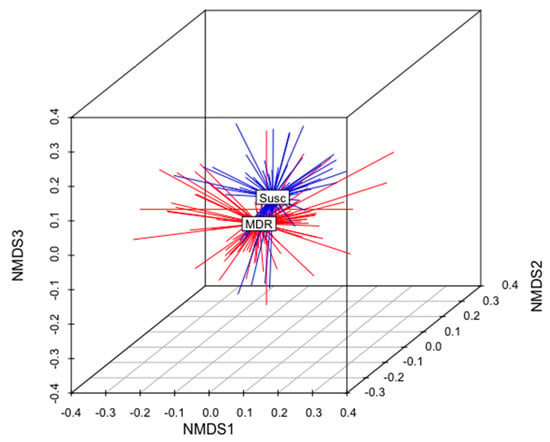
Figure 5.
Non-metric multidimensional scaling (NMDS) analysis of the presence and absence of genes in the genomes of all MDR and susceptible isolates in this analysis (k = 3; stress = 0.17; ANOSIM R = 0.16; p < 0.01). The centroid of each group (MDR or Susc) is labeled. MDR genomes are shown in red. S genomes are shown in blue.
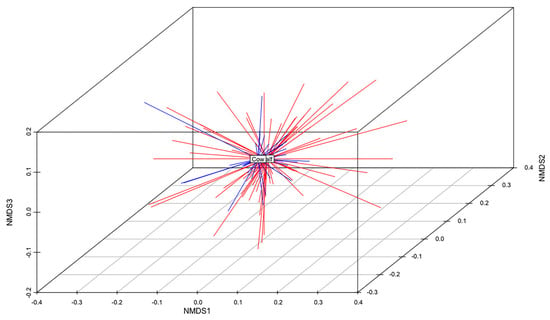
Figure 6.
Non-metric multidimensional scaling (NMDS) analysis of the presence and absence of genes in the genomes of all MDR isolates from calves and adult cows (k = 3; stress = 0.15; ANOSIM R = −0.08; p = 0.972). The centroid of each group (MDR calf or MDR cow) is labeled. MDR calf genomes are shown in red. MDR cow genomes are shown in blue.
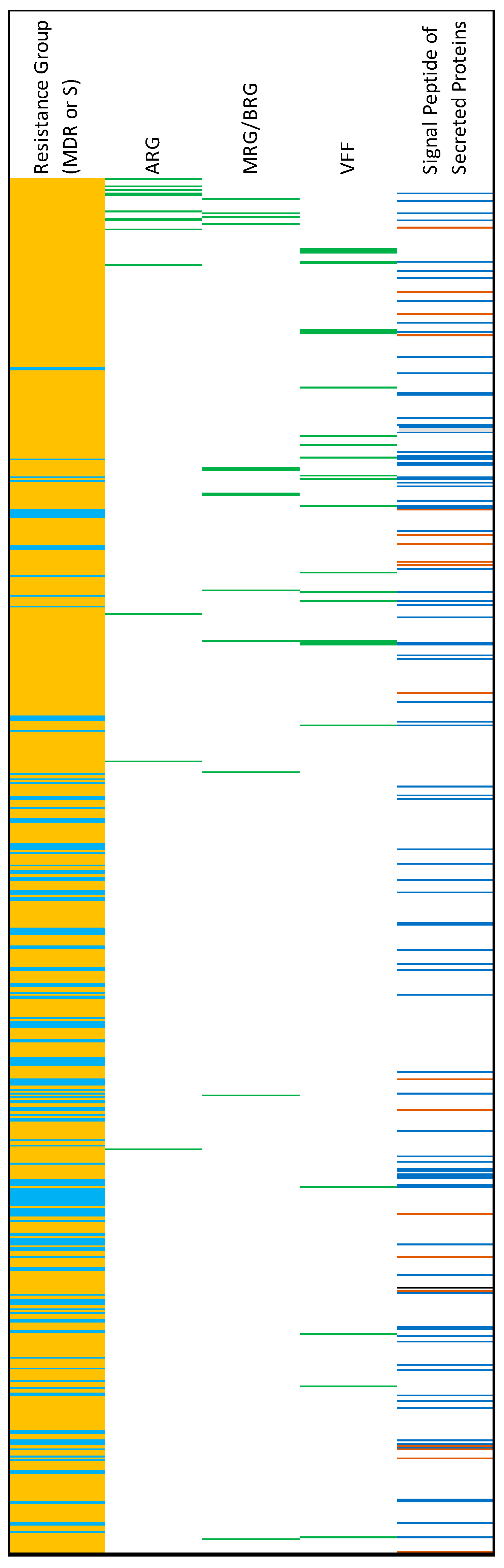
There was a total of 766 accessory genes that were significantly enriched in either MDR strains or in susceptible strains (Fisher’s Exact Test, q < 0.05) (Figure 7; Supplementary File S3). Of these, 619 (80%) were enriched in MDR strains, and 147 were enriched in susceptible strains. Thirteen of the 619 accessory genes that were enriched in MDR strains were identified as ARGs. Signal proteins (markers of secreted proteins) were detected in 86 protein products of the enriched genes in MDR strains (Figure 7; Supplementary File S3). When the enriched genes in the susceptible strains were translated, 22 signal proteins were detected.
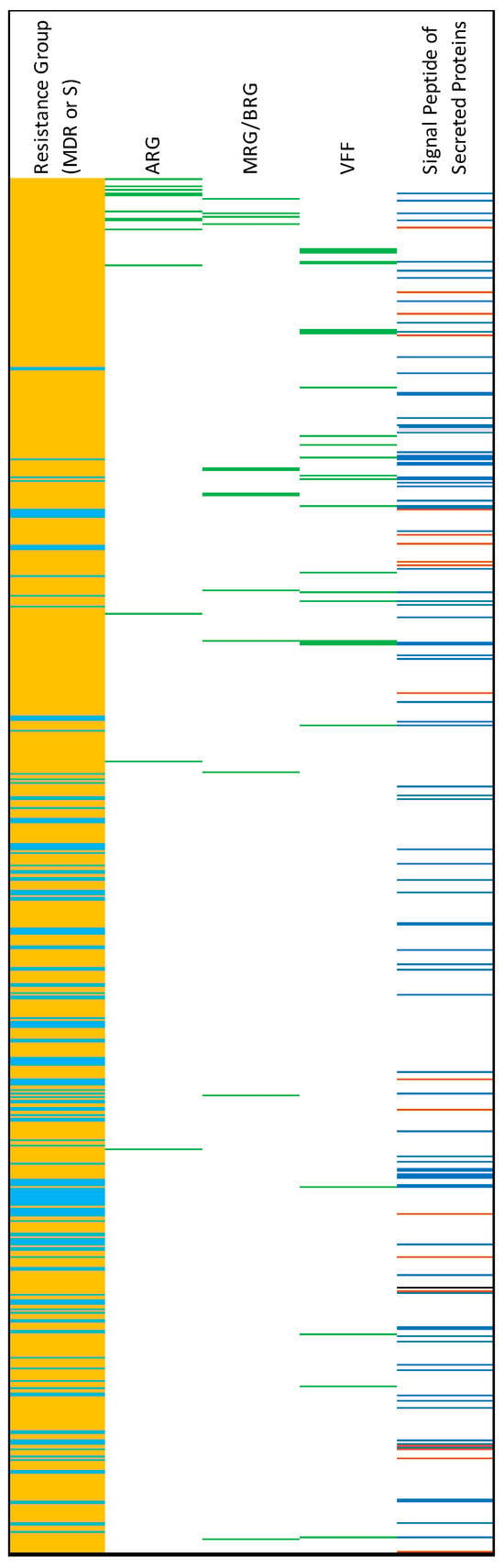
Figure 7.
Results of the Fisher’s Exact Test analysis of gene enrichment in MDR and susceptible (S) genomes. Light-blue on far-left column = genes enriched in S genomes. Orange = genes enriched in MDR genomes. For columns from 2 to 4: green = the corresponding gene is an antimicrobial resistance gene (ARG), metal resistance gene/biocide resistance gene (MRG/BRG), or virulence and fitness factor (VFF). For the last column, prediction of signal peptides of secreted proteins: blue = SP(Sec/SPI), brown = LIPO(Sec/SPII), black = TAT(Tat/SPI).
VFFs were also identified among the accessory genes enriched in either MDR or S genomes (Supplementary File S3). In the S genomes, these included cfaC (Mat/Ecp fimbriae outer membrane usher protein), cfaB (fimbrial protein), fimC (fimbrial chaperone protein), and fimI (long polar fimbria major subunit LpfA). In the MDR genomes, these included afa (adhesin), f17c-a (major type 1 subunit fimbrin), f17d-C (outer membrane usher protein), iuc-iutA (aerobactin synthesis and receptor), orgA (type III secretion apparatus protein OrgA/MxiK), pap (P-fimbriae), sit (iron and manganese transport), tia (Tia invasion determinant), and yjaa (YjaA family stress response protein).
Of the 619 genes enriched in MDR strains, 238 (38%) were annotated as known protein-coding genes. When assigned to KEGG functional categories, 26.8% were assigned to the signaling and cellular processes category of the protein families; 14.7% were assigned to the environmental information processing category; 13.4% were assigned to the unclassified genetic information processing category; 11.7% were assigned to the genetic information processing category of the protein families; 8.8% were assigned to the carbohydrate metabolism category, and 4.2% were assigned to the unclassified metabolism category.
Notable genes of interest known to confer significant phenotypes on cells that encode them that were enriched in the MDR genome include multiple iron acquisition systems, such as iucABCD-iutA (aerobactin synthesis and receptor), fecABCD (iron transport operon), sitACD (involved in iron and manganese transport), papABCD (P fimbriae), myo-inositol catabolism and transport genes iolABCDEG-iatA, and ascorbate metabolism genes ulaABC (Table 3). Multiple metal and biocide resistance genes were also enriched in MDR genomes. These include resistance to mercury (mer), copper (pco), silver (sil), and quaternary ammonia compounds (qacEΔ1) (Supplementary File S3).
Table 3.
Operons of interest that were enriched in MDR genomes. Also listed are the functions of these operons, the ranges of percent presence in MDR strains (not all genes within an operon were detected), maximum percent presence in susceptible strains, and the q-values indicating the statistical significance of their enrichment in MDR genomes (Fisher’s Exact Test).
4. Discussion
Results of this study demonstrate that genotypically antimicrobial-resistant and virulent E. coli are shed by dairy animals of all ages. Genes integral in the infection processes of all the major pathovars were detected among the strains. In previous studies, non-STEC strains isolated from bovine sources were frequently considered non-type-specific E. coli. However, a deeper genomic investigation of the strains in this study demonstrates that they often encode multiple VFFs as well as ARGs, demonstrating their human and animal health significance. Among these E. coli, STEC adulterant and ExPEC strains were repeatedly isolated. ExPEC strains are frequently MDR [35], and a high percentage of MDR strains in this study encoded ExPEC VFs, more so than the integral VFs of other pathovars.
Multiple ARGs conferring resistance to antimicrobials of human significance, particularly β-lactamases (blaCMY and blaCTX-M) and azithromycin (macrolide) resistance genes (mphA and mphB), along with point mutations conferring resistance to ciprofloxacin were identified in the genomes of E. coli isolated from the feces of dairy animals. β-lactams are among the most frequently used antibiotics in human clinical medicines, and resistance to these therapeutics has been increasing globally [36,37]. Fieldwork on these study farms demonstrated the presence of E. coli resistant to these antibiotics in the majority of farms, indicating that they are prevalent in dairy systems in the geographic study region [1]. Interestingly, blaCTX-M and, in particular, blaCMY were identified in more than half of MDR isolates, indicating that they were a significant component of MDR E. coli in these environments. This is further evidenced by the frequent co-occurrence of blaCMY with other ARGs in the E. coli genomes.
Based on an analysis of E. coli and the resistomes of calf feces, a higher ratio of resistant to susceptible bacteria is present in pre-weaned calf feces than in the feces of older animals [1,16]. Cao et al. [1] demonstrated this high level of resistance in pre- and post-weaned calves compared to dry or lactating cows, with pre-weaned calf feces harboring more resistant E. coli and more MDR E. coli than post-weaned calf feces. Previous studies have indicated that ARBs from teat surfaces can contaminate colostrum, which might seed the calf gut with ARBs, and that changes in diet may be, in part, responsible for the decrease in resistance abundance in these young animals. However, calves fed colostrum replacer and milk replacer similarly harbor high levels of resistance [38] that decrease over time, suggesting that there might be a selective pressure for ARBs in the calf gut that results in relatively high abundance of these organisms in young animals.
Previous work has shown that the presence of ARGs is associated with the presence of biocide and metal resistance genes (BRGs and MRGs) and that they are often collocated on the same plasmids or mobile elements [39,40]. It has been postulated that the presence of trace metals in feed selects ARBs that have MRGs encoded in their genomes [41,42]. Similar associations between ARGs, MRGs, and BRGs were identified in this study, most notably between the MDR genotype and the mercury resistance operon and the BRG, sugE1. However, the results of this study further demonstrate that E. coli with the MDR genotype is more likely to encode multiple suites of non-MRG/non-BRG genes encoding traits that may select for these organisms even in the absence of antimicrobial therapy or trace metal/biocide exposure and that these genes may point toward potential intervention strategies to reduce the carriage of ARBs in the herd or individual animal groups.
Our analyses indicated that the accessory gene contents of MDR isolates were somewhat different from those of the susceptible isolates, even when ARGs were removed from the analyses. Further, the gene content of MDR isolates from calves was not significantly different from that of adult cows, although the ratio of resistant to susceptible isolates is higher in calves than in cows [1]. Previous work has demonstrated that calves and adult cows can harbor the same MDR E. coli [43]. These data indicate that there are some genes that are more abundant in MDR strains than susceptible strains and that the gene contents of these MDR strains isolated from calves are approximately similar to those isolated from adult cows. Several of these genes compose operons that are involved in metabolic mechanisms that may be involved in this high MDR E. coli carriage in the calf gut. Of particular interest is the high number of genes enriched in the MDR isolates that are involved in iron acquisition. Iron is a limiting nutrient that is integral in many essential cellular processes. In human pathogenic E. coli, iron acquisition mechanisms are frequently found in ExPEC isolates, which are commensal in the gut but potentially pathogenic in the extraintestinal environment [44]. These systems are required for ExPEC colonization outside of the human gut, and studies have shown that they are upregulated in extraintestinal environments where available iron is limited [45,46,47]. However, the possible roles of iron acquisition mechanisms in the non-human mammalian gut have not been resolved. Iron scavenging via siderophores chelates iron, which transports these compounds back to the cell. Many taxa encode siderophores in the core chromosome, as well as accessory siderophores such as the sit system, aerobactin production, and salmochelin. The calf gut environment is relatively iron-depleted, and the main source of nutrition is usually colostrum for the first two days, followed by milk, both of which have a low iron content compared to that of the diets of older animals, which consume mostly forages and grains [48]. We hypothesize that the presence of siderophore systems (iucABCD-iutA and sitABCD), which are expressed in low-iron environments and are enriched in MDR strains in dairy animals, may allow for these strains to outcompete susceptible strains that typically lack these accessory iron scavenging systems. Siderophores are expressed in low-iron environments as a competitive mechanism to acquire this essential nutrient [49,50,51,52]. In E. coli, these genes (iucABCD-iutA and sitABCD) are typically encoded on incF plasmids, which also frequently encode ARGs, BRGs, and MRGs. We hypothesize that the collocation of plasmid-borne siderophores and ARGs selects for the latter in the low iron environment of the calf gut. Similar associations have been shown in E. coli collected from veal calves, which are typically fed an iron-limited diet [53].
The presence of other genes not involved in iron scavenging that are enriched in MDR strains suggests that the selection of ARGs within the calf gut may be multifactorial. These genes include myo-inositol transport and catabolism genes. Inositol is a carbocyclic sugar that has multiple structural and signaling roles in both eukaryotes and prokaryotes. It is found in both cow’s milk and feeds, such as cereal grains in its phosphorylated form, inositol hexakisphosphate (InsP6), or phytate, while forages have considerably lower InsP6 concentrations. Homologs of these ORFs have been identified in S. enterica, which can utilize inositol as a sole carbon source in vitro, indicating that it has an influence on the survival and, possibly, persistence of strains encoding these genes in environments with available inositol [54]. The introduction of forages and grains as a “calf starter” diet roughly coincides with a decrease in the carriage of MDR strains at around two weeks, which continues through the post-weaning period, suggesting that changes in diet may alter the abundance of resistance in the gut [2]. However, these genes are typically located on the E. coli chromosome, while most ARGs harbored by E. coli are plasmid-borne. It is not clear if there is a synergistic relationship between chromosomal-encoded genes and plasmid-borne ARGs; this association needs further evaluation.
Genes representing other, less defined mechanisms were also enriched in the MDR isolates; they may also confer a selective advantage for these bacteria in the calf gut and, hence, may also represent potential intervention targets for resistance in all animal groups. A significant number of genes were annotated as hypothetical proteins, indicating that their functions are not yet known. It may be that some of these are non-functional, but multiple hypothetical proteins were predicted to be translocated across the cell membrane, which suggests that they may be involved in interactions with the extracellular environment, which could include the bovine gastrointestinal tract. Further work identifying the functions of these genes in vitro and in vivo would need to be conducted to determine if they play any significant role in the survival of MDR strains in the animal gut.
MDR E. coli are frequently shed from calves and cows throughout the duration of their lives. The abundance of these resistant bacteria typically peaks at around two to three weeks of life, but it is still relatively high after weaning. The first two to three weeks after birth coincides with the time during which calves are primarily fed a milk diet, after which consumption of a calf starter, which has a higher concentration of iron than milk, occurs more frequently as the calf ages. These results demonstrate a need to further evaluate the role of iron availability within the calf gut as a potential driver of the high abundance of AMR in the feces of young calves, which may reveal a relatively simple approach to reducing the carriage of MDR E. coli in the dairy calf gut. Such an approach could have One Health implications by mitigating or disrupting the flow of multidrug-resistant E. coli between dairy farms, animals, and the environment.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/antibiotics12101559/s1, Supplementary File S1: Co-occurrence results between antimicrobial-resistance genes (ARGs), biocide resistance genes (BRGs), and metal resistance genes (MRGs). Supplementary File S2: Full virulence matrix of all study strains is used in this analysis. Supplementary File S3: Genes enriched in MDR and susceptible genes along with their corresponding E. coli NCBI protein ID, BRG/MRG, annotation, SignalP identity, and KEGG orthology (KO). Supplementary File S4: NCBI accession numbers of E. coli genomes sequenced for this study.
Author Contributions
B.J.H. designed and coordinated the study, analyzed the data, and prepared the manuscript; S.S. analyzed data; S.W.K. carried out sequencing and analyzed data; E.H. collected samples; J.A.S.V.K. edited and critically revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This project was funded by internal USDA-ARS funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All genome sequencing data generated in this study are publicly available at NCBI (Supplementary File S4).
Acknowledgments
We would like to thank Jakeitha Sonnier and Laura Del Collo for their assistance with laboratory work. Mention of trade names or commercial products in this article is solely for providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cao, H.; Pradhan, A.K.; Karns, J.S.; Hovingh, E.; Wolfgang, D.R.; Vinyard, B.T.; Kim, S.W.; Salaheen, S.; Haley, B.J.; Van Kessel, J.A.S. Age-Associated Distribution of Antimicrobial-Resistant Salmonella enterica and Escherichia coli Isolated from Dairy Herds in Pennsylvania, 2013–2015. Foodborne Pathog. Dis. 2019, 16, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Taft, D.H.; Maldonado-Gomez, M.X.; Johnson, D.; Treiber, M.L.; Lemay, D.G.; DePeters, E.J.; Mills, D.A. The fecal resistome of dairy cattle is associated with diet during nursing. Nat. Commun. 2019, 10, 4406. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, Y.; Jay-Russell, M.; Lemay, D.G.; Mills, D.A. Reservoirs of antimicrobial resistance genes in retail raw milk. Microbiome 2020, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Gyles, C.L. Shiga toxin-producing Escherichia coli: An overview. J. Anim. Sci. 2007, 85, E45–E62. [Google Scholar] [CrossRef] [PubMed]
- Salaheen, S.; Kim, S.W.; Springer, H.R.; Hovingh, E.P.; Van Kessel, J.A.S.; Haley, B.J. Genomic diversity of antimicrobial-resistant and Shiga toxin gene-harboring non-O157 Escherichia coli from dairy calves. J. Glob. Antimicrob. Resist. 2023, 33, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L.T. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef]
- Call, D.R.; Davis, M.A.; Sawant, A.A. Antimicrobial resistance in beef and dairy cattle production. Anim. Health Res. Rev. 2008, 9, 159–167. [Google Scholar] [CrossRef]
- Springer, H.R.; Denagamage, T.N.; Fenton, G.D.; Haley, B.J.; Van Kessel, J.A.S.; Hovingh, E.P. Antimicrobial Resistance in Fecal Escherichia coli and Salmonella enterica from Dairy Calves: A Systematic Review. Foodborne Pathog. Dis. 2019, 16, 23–34. [Google Scholar] [CrossRef]
- Durso, L.M.; Harhay, G.P.; Bono, J.L.; Smith, T.P. Virulence-associated and antibiotic resistance genes of microbial populations in cattle feces analyzed using a metagenomic approach. J. Microbiol. Methods 2011, 84, 278–282. [Google Scholar] [CrossRef]
- Wichmann, F.; Udikovic-Kolic, N.; Andrew, S.; Handelsman, J. Diverse Antibiotic Resistance Genes in Dairy Cow Manure. mBio 2014, 5, e01017. [Google Scholar] [CrossRef]
- Haley, B.J.; Van Kessel, J.A.S. The resistome of the bovine gastrointestinal tract. Curr. Opin. Biotechnol. 2022, 73, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Salaheen, S.; Kim, S.W.; Springer, H.R.; Hovingh, E.P.; Van Kessel, J.A.S.; Haley, B.J. Characterization of Antimicrobial Resistance Genes and Virulence Factors in the Genomes of Escherichia coli ST69 Isolates from Preweaned Dairy Calves and Their Phylogenetic Relationship with Poultry and Human Clinical Strains. Microb. Drug Resist. 2023, 29, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Schmidt, J.W. FunctionalblaKPC-2Sequences Are Present in U.S. Beef Cattle Feces Regardless of Antibiotic Use. Foodborne Pathog. Dis. 2018, 15, 444–448. [Google Scholar] [CrossRef]
- Noyes, N.R.; Yang, X.; Linke, L.M.; Magnuson, R.J.; Cook, S.R.; Zaheer, R.; Yang, H.; Woerner, D.R.; Geornaras, I.; McArt, J.A.; et al. Characterization of the resistome in manure, soil and wastewater from dairy and beef production systems. Sci. Rep. 2016, 6, 24645. [Google Scholar] [CrossRef] [PubMed]
- Rovira, P.; McAllister, T.; Lakin, S.M.; Cook, S.R.; Doster, E.; Noyes, N.R.; Weinroth, M.D.; Yang, X.; Parker, J.K.; Boucher, C.; et al. Characterization of the Microbial Resistome in Conventional and “Raised Without Antibiotics” Beef and Dairy Production Systems. Front. Microbiol. 2019, 10, 1980. [Google Scholar] [CrossRef]
- Haley, B.J.; Kim, S.-W.; Salaheen, S.; Hovingh, E.; Van Kessel, J.A.S. Differences in the Microbial Community and Resistome Structures of Feces from Preweaned Calves and Lactating Dairy Cows in Commercial Dairy Herds. Foodborne Pathog. Dis. 2020, 17, 494–503. [Google Scholar] [CrossRef]
- Gaire, T.N.; Scott, H.M.; Sellers, L.; Nagaraja, T.G.; Volkova, V.V. Age Dependence of Antimicrobial Resistance Among Fecal Bacteria in Animals: A Scoping Review. Front. Veter. Sci. 2021, 7, 622495. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontéen, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Wirth, T.; Falush, D.; Lan, R.; Colles, F.; Mensa, P.; Wieler, L.H.; Karch, H.; Reeves, P.R.; Maiden, M.C.J.; Ochman, H.; et al. Sex and virulence in Escherichia coli: An evolutionary perspective. Mol. Microbiol. 2006, 60, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; Garcìa-Fernandez, A.; Larsen, M.; Lund, O.; Voldby Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids. Antimicrob using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.-F.; Mohamed, K.; Fan, Y.; Achtman, M.; the Agama Study Group; Brown, D.; Chattaway, M.; Dallman, T.; Delahay, R.; et al. The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Abricate, Github. 2020. Available online: https://github.com/tseemann/abricate (accessed on 28 March 2023).
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Food Safety and Inspection Service, Federal Register. Vol. 77, No. 105 Thursday, 31 May 2012. Available online: https://www.govinfo.gov/content/pkg/FR-2012-05-31/pdf/FR-2012-05-31.pdf (accessed on 2 April 2023).
- Spurbeck, R.R.; Dinh, P.C., Jr.; Walk, S.T.; Stapleton, A.E.; Hooton, T.M.; Nolan, L.K.; Kim, K.S.; Johnson, J.R.; Mobley, H.L.T. Escherichia coli Isolates That Carry vat, fyuA, chuA, and yfcV Efficiently Colonize the Urinary Tract. Infect. Immun. 2012, 80, 4115–4122. [Google Scholar] [CrossRef]
- Mellata, M.; Ahmed, L.N.; Price, L.B.; Graham, J.P.; Gazal, L.E.S.; Puño-Sarmiento, J.J.; Medeiros, L.P.; Cyoia, P.S.; da Silveira, W.D.; Nakazato, G.; et al. Human and Avian Extraintestinal Pathogenic Escherichia coli: Infections, Zoonotic Risks, and Antibiotic Resistance Trends. Foodborne Pathog. Dis. 2013, 10, 916–932. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.Y.; Van Boeckel, T.P.; Martinez, E.M.; Pant, S.; Gandra, S.; Levin, S.A.; Goossens, H.; Laxminarayan, R. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. USA 2018, 115, E3463–E3470. [Google Scholar] [CrossRef] [PubMed]
- Springer, H.R.; Van Kessel, J.S.; Haley, B.J.; Kim, S.W.; Vinyard, B.; Hovingh, E. Impact of Weaning Age and Antibiotic Residues in the Diet on Dynamics of Antimicrobial-Resistant Fecal Escherichia coli in Dairy Calves; United States Department of Agriculture: Beltsville, MD, USA, 2023; to be submitted. [Google Scholar]
- Li, L.-G.; Xia, Y.; Zhang, T. Co-occurrence of antibiotic and metal resistance genes revealed in complete genome collection. ISME J. 2017, 11, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.C.; Oladeinde, A.; Kieran, T.J.; Finger, J.W.; Bayona-Vásquez, N.J.; Cartee, J.C.; Beasley, J.C.; Seaman, J.C.; McArthur, J.V.; Rhodes, O.E.; et al. Co-occurrence of antibiotic, biocide, and heavy metal resistance genes in bacteria from metal and radionuclide contaminated soils at the Savannah River Site. Microb. Biotechnol. 2020, 13, 1179–1200. [Google Scholar] [CrossRef]
- Medardus, J.J.; Molla, B.Z.; Nicol, M.; Morrow, W.M.; Rajala-Schultz, P.J.; Kazwala, R.; Gebreyes, W.A. In-Feed Use of Heavy Metal Micronutrients in U.S. Swine Production Systems and Its Role in Persistence of Multidrug-Resistant Salmonellae. Appl. Environ. Microbiol. 2014, 80, 2317–2325. [Google Scholar] [CrossRef]
- Bearson, B.L.; Trachsel, J.M.; Shippy, D.C.; Sivasankaran, S.K.; Kerr, B.J.; Loving, C.L.; Brunelle, B.W.; Curry, S.M.; Gabler, N.K.; Bearson, S.M.D. The Role of Salmonella Genomic Island 4 in Metal Tolerance of Salmonella enterica Serovar I 4,[5],12:i:-Pork Outbreak Isolate USDA15WA-1. Genes 2020, 11, 1291. [Google Scholar] [CrossRef]
- Salaheen, S.; Cao, H.; Sonnier, J.L.; Kim, S.W.; Del Collo, L.P.; Hovingh, E.; Karns, J.S.; Haley, B.J.; Van Kessel, J.A.S. Diversity of Extended-Spectrum Cephalosporin-Resistant Escherichia coli in Feces from Calves and Cows on Pennsylvania Dairy Farms. Foodborne Pathog. Dis. 2019, 16, 368–370. [Google Scholar] [CrossRef]
- Dale, A.P.; Woodford, N. Extra-intestinal pathogenic Escherichia coli (ExPEC): Disease, carriage and clones. J. Infect. 2015, 71, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Roos, V.; Ulett, G.C.; Schembri, M.A.; Klemm, P. The Asymptomatic Bacteriuria Escherichia coli Strain 83972 Outcompetes Uropathogenic E. coli Strains in Human Urine. Infect. Immun. 2006, 74, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Hagan, E.C.; Mobley, H.L.T. Haem acquisition is facilitated by a novel receptor Hma and required by uropathogenic Escherichia coli for kidney infection. Mol. Microbiol. 2009, 71, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Skyberg, J.A.; Johnson, T.J.; Nolan, L.K. Mutational and transcriptional analyses of an avian pathogenic Escherichia coli ColV plasmid. BMC Microbiol. 2008, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.; Pinos-Rodríguez, J.; Socha, M. Effects of feeding supplemental organic iron to late gestation and early lactation dairy cows. J. Dairy Sci. 2010, 93, 2153–2160. [Google Scholar] [CrossRef]
- Miethke, M.; Marahiel, M.A. Siderophore-Based Iron Acquisition and Pathogen Control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef]
- Chu, B.C.; Garcia-Herrero, A.; Johanson, T.H.; Krewulak, K.D.; Lau, C.K.; Peacock, R.S.; Slavinskaya, Z.; Vogel, H.J. Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. BioMetals 2010, 23, 601–611. [Google Scholar] [CrossRef]
- Ahmed, E.; Holmström, S.J.M. Siderophores in environmental research: Roles and applications. Microb. Biotechnol. 2014, 7, 196–208. [Google Scholar] [CrossRef]
- Garénaux, A.; Caza, M.; Dozois, C.M. The Ins and Outs of siderophore mediated iron uptake by extra-intestinal pathogenic Escherichia coli. Veter. Microbiol. 2011, 153, 89–98. [Google Scholar] [CrossRef]
- Haley, B.J.; Kim, S.W.; Salaheen, S.; Hovingh, E.; Van Kessel, J.A.S. Virulome and genome analyses identify associations between antimicrobial resistance genes and virulence factors in highly drug-resistant Escherichia coli isolated from veal calves. PLoS ONE 2022, 17, e0265445. [Google Scholar] [CrossRef]
- Kröger, C.; Fuchs, T.M. Characterization of the myo -Inositol Utilization Island of Salmonella enterica serovar Typhimurium. J. Bacteriol. 2009, 191, 545–554. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).