

Impacts of Hydrophobic Mismatch on Antimicrobial Peptide Efficacy and Bilayer Permeabilization



Abstract
:1. Introduction
2. Results
2.1. Peptide Composition
2.2. Antimicrobial Activity
2.3. Binding Assays
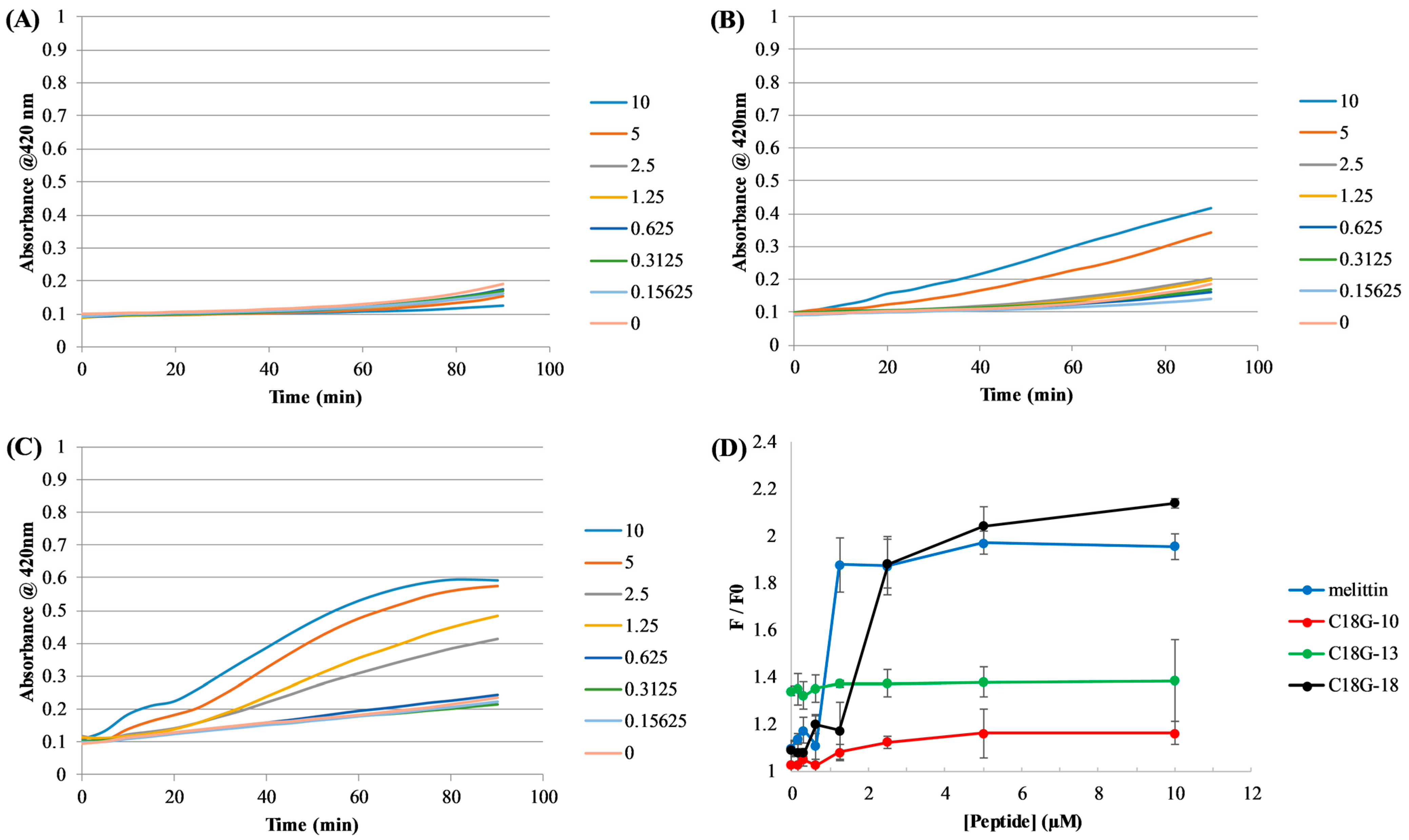
2.4. Bacterial Membrane Permeabilization
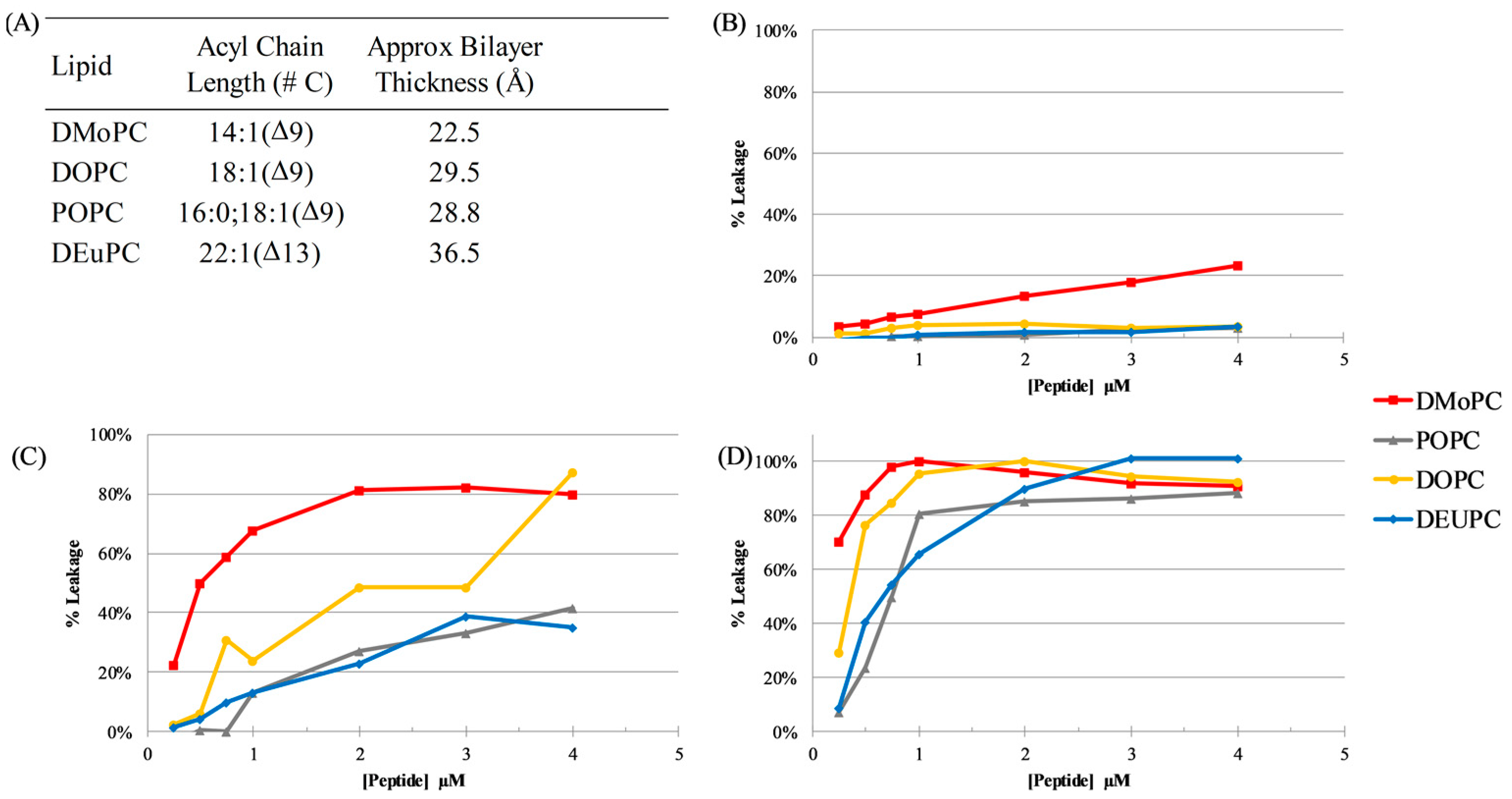
2.5. Vesicle Permeabilization
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Bacterial Strains and Culture
4.3. Minimal Inhibitory Concentration Assay
4.4. Lipid Binding Assays
4.5. Bacterial Membrane Permeabilization
4.6. Vesicle Leakage Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- CDC. Antibiotic Resistance Threats in the United States, 2019; U.S. Department of Health and Human Services, CDC: Washington, DC, USA, 2019.
- Ahmad, M.; Khan, A.U. Global economic impact of antibiotic resistance: A review. J. Glob. Antimicrob. Resist. 2019, 19, 313–316. [Google Scholar] [CrossRef]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Seethaler, M.; Hertlein, T.; Wecklein, B.; Ymeraj, A.; Ohlsen, K.; Lalk, M.; Hilgeroth, A. Novel Small-molecule Antibacterials against Gram-positive Pathogens of Staphylococcus and Enterococcus Species. Antibiotics 2019, 8, 210. [Google Scholar] [CrossRef]
- Iacopetta, D.; Ceramella, J.; Catalano, A.; D’Amato, A.; Lauria, G.; Saturnino, C.; Andreu, I.; Longo, P.; Sinicropi, M.S. Diarylureas: New Promising Small Molecules against Streptococcus mutans for the Treatment of Dental Caries. Antibiotics 2023, 12, 112. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M. Antimicrobial Peptides: From Design to Clinical Application. Antibiotics 2022, 11, 349. [Google Scholar] [CrossRef]
- Makhlynets, O.V.; Caputo, G.A. Characteristics and therapeutic applications of antimicrobial peptides. Biophys. Rev. 2021, 2, 011301. [Google Scholar] [CrossRef]
- Teng, P.; Shao, H.; Huang, B.; Xie, J.; Cui, S.; Wang, K.; Cai, J. Small Molecular Mimetics of Antimicrobial Peptides as a Promising Therapy To Combat Bacterial Resistance. J. Med. Chem. 2023, 66, 2211–2234. [Google Scholar] [CrossRef]
- Yasuhara, K.; Tsukamoto, M.; Kikuchi, J.I.; Kuroda, K. An Antimicrobial Peptide-Mimetic Methacrylate Random Copolymer Induces Domain Formation in a Model Bacterial Membrane. J. Membr. Biol. 2022, 255, 513–521. [Google Scholar] [CrossRef]
- Panjla, A.; Kaul, G.; Chopra, S.; Titz, A.; Verma, S. Short Peptides and Their Mimetics as Potent Antibacterial Agents and Antibiotic Adjuvants. ACS Chem. Biol. 2021, 16, 2731–2745. [Google Scholar] [CrossRef]
- Mankoci, S.; Ewing, J.; Dalai, P.; Sahai, N.; Barton, H.A.; Joy, A. Bacterial Membrane Selective Antimicrobial Peptide-Mimetic Polyurethanes: Structure-Property Correlations and Mechanisms of Action. Biomacromolecules 2019, 20, 4096–4106. [Google Scholar] [CrossRef]
- Dos Reis, T.F.; de Castro, P.A.; Bastos, R.W.; Pinzan, C.F.; Souza, P.F.N.; Ackloo, S.; Hossain, M.A.; Drewry, D.H.; Alkhazraji, S.; Ibrahim, A.S.; et al. A host defense peptide mimetic, brilacidin, potentiates caspofungin antifungal activity against human pathogenic fungi. Nat. Commun. 2023, 14, 2052. [Google Scholar] [CrossRef]
- D’Souza, A.; Yoon, J.H.; Beaman, H.; Gosavi, P.; Lengyel-Zhand, Z.; Sternisha, A.; Centola, G.; Marshall, L.R.; Wehrman, M.D.; Schultz, K.M.; et al. Nine-Residue Peptide Self-Assembles in the Presence of Silver to Produce a Self-Healing, Cytocompatible, Antimicrobial Hydrogel. ACS Appl. Mater. Interfaces 2020, 12, 17091–17099. [Google Scholar] [CrossRef]
- Edirisinghe, D.I.U.; D’Souza, A.; Ramezani, M.; Carroll, R.J.; Chicon, Q.; Muenzel, C.L.; Soule, J.; Monroe, M.B.B.; Patteson, A.E.; Makhlynets, O.V. Antibacterial and Cytocompatible pH-Responsive Peptide Hydrogel. Molecules 2023, 28, 4390. [Google Scholar] [CrossRef]
- Palermo, E.F.; Kuroda, K. Structural determinants of antimicrobial activity in polymers which mimic host defense peptides. Appl. Microbiol. Biotechnol. 2010, 87, 1605–1615. [Google Scholar] [CrossRef]
- Jones, J.B.; Liu, L.; Rank, L.A.; Wetzel, D.; Woods, E.C.; Biok, N.; Anderson, S.E.; Lee, M.R.; Liu, R.; Huth, S.; et al. Cationic Homopolymers Inhibit Spore and Vegetative Cell Growth of Clostridioides difficile. ACS Infect. Dis. 2021, 7, 1236–1247. [Google Scholar] [CrossRef]
- Brackman, G.; Coenye, T. Quorum sensing inhibitors as anti-biofilm agents. Curr. Pharm. Des. 2015, 21, 5–11. [Google Scholar] [CrossRef]
- Wu, B.; Capilato, J.; Pham, M.P.; Walker, J.; Spur, B.; Rodriguez, A.; Perez, L.J.; Yin, K. Lipoxin A4 augments host defense in sepsis and reduces Pseudomonas aeruginosa virulence through quorum sensing inhibition. FASEB J. 2016, 30, 2400–2410. [Google Scholar] [CrossRef]
- Goderecci, S.S.; Kaiser, E.; Yanakas, M.; Norris, Z.; Scaturro, J.; Oszust, R.; Medina, C.D.; Waechter, F.; Heon, M.; Krchnavek, R.R.; et al. Silver Oxide Coatings with High Silver-Ion Elution Rates and Characterization of Bactericidal Activity. Molecules 2017, 22, 1487. [Google Scholar] [CrossRef]
- Grass, G.; Rensing, C.; Solioz, M. Metallic copper as an antimicrobial surface. Appl. Environ. Microbiol. 2011, 77, 1541–1547. [Google Scholar] [CrossRef]
- Pasquet, J.; Chevalier, Y.; Pelletier, J.; Couval, E.; Bouvier, D.; Bolzinger, M.-A. The contribution of zinc ions to the antimicrobial activity of zinc oxide. Colloids Surf. A Physicochem. Eng. Asp. 2014, 457, 263–274. [Google Scholar] [CrossRef]
- Sharmin, S.; Rahaman, M.M.; Sarkar, C.; Atolani, O.; Islam, M.T.; Adeyemi, O.S. Nanoparticles as antimicrobial and antiviral agents: A literature-based perspective study. Heliyon 2021, 7, e06456. [Google Scholar] [CrossRef]
- Urnukhsaikhan, E.; Bold, B.E.; Gunbileg, A.; Sukhbaatar, N.; Mishig-Ochir, T. Antibacterial activity and characteristics of silver nanoparticles biosynthesized from Carduus crispus. Sci. Rep. 2021, 11, 21047. [Google Scholar] [CrossRef]
- MacNair, C.R.; Tsai, C.N.; Rutherford, S.T.; Tan, M.W. Returning to Nature for the Next Generation of Antimicrobial Therapeutics. Antibiotics 2023, 12, 1267. [Google Scholar] [CrossRef]
- Palla, F.; Bucchini, A.E.A.; Giamperi, L.; Marino, P.; Raimondo, F.M. Plant Extracts as Antimicrobial Agents in Sustainable Conservation of Erythrina caffra (Fabaceae) Historical Trees. Antibiotics 2023, 12, 1098. [Google Scholar] [CrossRef]
- Kvich, L.; Christensen, M.H.; Pierchala, M.K.; Astafiev, K.; Lou-Moeller, R.; Bjarnsholt, T. The Combination of Low-Frequency Ultrasound and Antibiotics Improves the Killing of In Vitro Staphylococcus aureus and Pseudomonas aeruginosa Biofilms. Antibiotics 2022, 11, 1494. [Google Scholar] [CrossRef]
- Sousa, M.; Afonso, A.C.; Teixeira, L.S.; Borges, A.; Saavedra, M.J.; Simoes, L.C.; Simoes, M. Hydrocinnamic Acid and Perillyl Alcohol Potentiate the Action of Antibiotics against Escherichia coli. Antibiotics 2023, 12, 360. [Google Scholar] [CrossRef]
- Yang, D.D.; Paterna, N.J.; Senetra, A.S.; Casey, K.R.; Trieu, P.D.; Caputo, G.A.; Vaden, T.D.; Carone, B.R. Synergistic interactions of ionic liquids and antimicrobials improve drug efficacy. iScience 2021, 24, 101853. [Google Scholar] [CrossRef]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Lu, W. Antimicrobial peptides. Semin. Cell Dev. Biol. 2019, 88, 105–106. [Google Scholar] [CrossRef]
- Luo, Y.; Song, Y. Mechanism of Antimicrobial Peptides: Antimicrobial, Anti-Inflammatory and Antibiofilm Activities. Int. J. Mol. Sci. 2021, 22, 11401. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, L.; Dong, C. Antimicrobial Peptides: An Overview of their Structure, Function and Mechanism of Action. Protein Pept. Lett. 2022, 29, 641–650. [Google Scholar] [CrossRef]
- Dijksteel, G.S.; Ulrich, M.M.W.; Middelkoop, E.; Boekema, B. Review: Lessons Learned From Clinical Trials Using Antimicrobial Peptides (AMPs). Front. Microbiol. 2021, 12, 616979. [Google Scholar] [CrossRef]
- Darveau, R.P.; Blake, J.; Seachord, C.L.; Cosand, W.L.; Cunningham, M.D.; Cassiano-Clough, L.; Maloney, G. Peptides related to the carboxyl terminus of human platelet factor IV with antibacterial activity. J. Clin. Investig. 1992, 90, 447–455. [Google Scholar] [CrossRef]
- Peck-Miller, K.A.; Blake, J.; Cosand, W.L.; Darveau, R.P.; Fell, H.P. Structure-activity analysis of the antitumor and hemolytic properties of the amphiphilic alpha-helical peptide, C18G. Int. J. Pept. Protein. Res. 1994, 44, 143–151. [Google Scholar] [CrossRef]
- Yadavalli, S.S.; Carey, J.N.; Leibman, R.S.; Chen, A.I.; Stern, A.M.; Roggiani, M.; Lippa, A.M.; Goulian, M. Antimicrobial peptides trigger a division block in Escherichia coli through stimulation of a signalling system. Nat. Commun. 2016, 7, 12340. [Google Scholar] [CrossRef]
- Choi, J.; Groisman, E.A. Activation of master virulence regulator PhoP in acidic pH requires the Salmonella-specific protein UgtL. Sci. Signal. 2017, 10, eaan6284. [Google Scholar] [CrossRef]
- Moskowitz, S.M.; Ernst, R.K.; Miller, S.I. PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J. Bacteriol. 2004, 186, 575–579. [Google Scholar] [CrossRef]
- Hitchner, M.A.; Necelis, M.R.; Shirley, D.; Caputo, G.A. Effect of Non-natural Hydrophobic Amino Acids on the Efficacy and Properties of the Antimicrobial Peptide C18G. Probiotics Antimicrob. Proteins 2020, 13, 527–541. [Google Scholar] [CrossRef]
- Kohn, E.M.; Shirley, D.J.; Arotsky, L.; Picciano, A.M.; Ridgway, Z.; Urban, M.W.; Carone, B.R.; Caputo, G.A. Role of Cationic Side Chains in the Antimicrobial Activity of C18G. Molecules 2018, 23, 329. [Google Scholar] [CrossRef]
- Saint Jean, K.D.; Henderson, K.D.; Chrom, C.L.; Abiuso, L.E.; Renn, L.M.; Caputo, G.A. Effects of Hydrophobic Amino Acid Substitutions on Antimicrobial Peptide Behavior. Probiotics Antimicrob. Proteins 2018, 10, 408–419. [Google Scholar] [CrossRef]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef]
- Hristova, K.; White, S.H. An experiment-based algorithm for predicting the partitioning of unfolded peptides into phosphatidylcholine bilayer interfaces. Biochemistry 2005, 44, 12614–12619. [Google Scholar] [CrossRef]
- Walker, J.M. The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Ramsey, J.; Rasche, H.; Maughmer, C.; Criscione, A.; Mijalis, E.; Liu, M.; Hu, J.C.; Young, R.; Gill, J.J. Galaxy and Apollo as a biologist-friendly interface for high-quality cooperative phage genome annotation. PLoS Comput. Biol. 2020, 16, e1008214. [Google Scholar] [CrossRef]
- Raghuraman, H.; Chatterjee, S.; Das, A. Site-Directed Fluorescence Approaches for Dynamic Structural Biology of Membrane Peptides and Proteins. Front. Mol. Biosci. 2019, 6, 96. [Google Scholar] [CrossRef]
- Raghuraman, H.; Chattopadhyay, A. Interaction of melittin with membrane cholesterol: A fluorescence approach. Biophys. J. 2004, 87, 2419–2432. [Google Scholar] [CrossRef]
- Ridgway, Z.; Picciano, A.L.; Gosavi, P.M.; Moroz, Y.S.; Angevine, C.E.; Chavis, A.E.; Reiner, J.E.; Korendovych, I.V.; Caputo, G.A. Functional characterization of a melittin analog containing a non-natural tryptophan analog. Biopolymers 2015, 104, 384–394. [Google Scholar] [CrossRef]
- Sovadinova, I.; Palermo, E.F.; Huang, R.; Thoma, L.M.; Kuroda, K. Mechanism of polymer-induced hemolysis: Nanosized pore formation and osmotic lysis. Biomacromolecules 2011, 12, 260–268. [Google Scholar] [CrossRef]
- Hills, R.D., Jr.; McGlinchey, N. Model parameters for simulation of physiological lipids. J. Comput. Chem. 2016, 37, 1112–1118. [Google Scholar] [CrossRef]
- Ridder, A.N.; van de Hoef, W.; Stam, J.; Kuhn, A.; de Kruijff, B.; Killian, J.A. Importance of hydrophobic matching for spontaneous insertion of a single-spanning membrane protein. Biochemistry 2002, 41, 4946–4952. [Google Scholar] [CrossRef]
- Killian, J.A. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta 1998, 1376, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Foo, A.C.; Harvey, B.G.; Metz, J.J.; Goto, N.K. Influence of hydrophobic mismatch on the catalytic activity of Escherichia coli GlpG rhomboid protease. Protein Sci. 2015, 24, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Soubias, O.; Niu, S.L.; Mitchell, D.C.; Gawrisch, K. Lipid-rhodopsin hydrophobic mismatch alters rhodopsin helical content. J. Am. Chem. Soc. 2008, 130, 12465–12471. [Google Scholar] [CrossRef]
- Windisch, D.; Ziegler, C.; Grage, S.L.; Burck, J.; Zeitler, M.; Gor’kov, P.L.; Ulrich, A.S. Hydrophobic Mismatch Drives the Interaction of E5 with the Transmembrane Segment of PDGF Receptor. Biophys. J. 2015, 109, 737–749. [Google Scholar] [CrossRef]
- Caputo, G.A.; London, E. Cumulative effects of amino acid substitutions and hydrophobic mismatch upon the transmembrane stability and conformation of hydrophobic alpha-helices. Biochemistry 2003, 42, 3275–3285. [Google Scholar] [CrossRef]
- Lin, Q.; London, E. Altering hydrophobic sequence lengths shows that hydrophobic mismatch controls affinity for ordered lipid domains (rafts) in the multitransmembrane strand protein perfringolysin O. J. Biol. Chem. 2013, 288, 1340–1352. [Google Scholar] [CrossRef]
- Milovanovic, D.; Honigmann, A.; Koike, S.; Gottfert, F.; Pahler, G.; Junius, M.; Mullar, S.; Diederichsen, U.; Janshoff, A.; Grubmuller, H.; et al. Hydrophobic mismatch sorts SNARE proteins into distinct membrane domains. Nat. Commun. 2015, 6, 5984. [Google Scholar] [CrossRef] [PubMed]
- Monne, M.; von Heijne, G. Effects of ‘hydrophobic mismatch’ on the location of transmembrane helices in the ER membrane. FEBS Lett. 2001, 496, 96–100. [Google Scholar] [CrossRef]
- Schmidt, U.; Weiss, M. Hydrophobic mismatch-induced clustering as a primer for protein sorting in the secretory pathway. Biophys. Chem. 2010, 151, 34–38. [Google Scholar] [CrossRef]
- Basu, I.; Chattopadhyay, A.; Mukhopadhyay, C. Ion channel stability of Gramicidin A in lipid bilayers: Effect of hydrophobic mismatch. Biochim. Biophys. Acta 2014, 1838, 328–338. [Google Scholar] [CrossRef]
- Hao, B.; Zhou, W.; Theg, S.M. Hydrophobic mismatch is a key factor in protein transport across lipid bilayer membranes via the Tat pathway. J. Biol. Chem. 2022, 298, 101991. [Google Scholar] [CrossRef]
- Mehner-Breitfeld, D.; Ringel, M.T.; Tichy, D.A.; Endter, L.J.; Stroh, K.S.; Lunsdorf, H.; Risselada, H.J.; Bruser, T. TatA and TatB generate a hydrophobic mismatch important for the function and assembly of the Tat translocon in Escherichia coli. J. Biol. Chem. 2022, 298, 102236. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, D.A.; Chattopadhyay, A. Modulation of gramicidin channel conformation and organization by hydrophobic mismatch in saturated phosphatidylcholine bilayers. Biochim. Biophys. Acta 2007, 1768, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tristram-Nagle, S.; Nagle, J.F. Alamethicin aggregation in lipid membranes. J. Membr. Biol. 2009, 231, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Lee, K.I.; Morris, P.; Pastor, R.W.; Andersen, O.S.; Im, W. Influence of hydrophobic mismatch on structures and dynamics of gramicidin a and lipid bilayers. Biophys. J. 2012, 102, 1551–1560. [Google Scholar] [CrossRef]
- Rui, H.; Im, W. Protegrin-1 orientation and physicochemical properties in membrane bilayers studied by potential of mean force calculations. J. Comput. Chem. 2010, 31, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.C.; Strandberg, E.; Grau-Campistany, A.; Wadhwani, P.; Reichert, J.; Burck, J.; Rabanal, F.; Auger, M.; Paquin, J.F.; Ulrich, A.S. Influence of the Length and Charge on the Activity of alpha-Helical Amphipathic Antimicrobial Peptides. Biochemistry 2017, 56, 1680–1695. [Google Scholar] [CrossRef] [PubMed]
- Grau-Campistany, A.; Strandberg, E.; Wadhwani, P.; Rabanal, F.; Ulrich, A.S. Extending the Hydrophobic Mismatch Concept to Amphiphilic Membranolytic Peptides. J. Phys. Chem. Lett. 2016, 7, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Grau-Campistany, A.; Strandberg, E.; Wadhwani, P.; Reichert, J.; Burck, J.; Rabanal, F.; Ulrich, A.S. Hydrophobic mismatch demonstrated for membranolytic peptides, and their use as molecular rulers to measure bilayer thickness in native cells. Sci. Rep. 2015, 5, 9388. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Prediction of the secondary structure of proteins from their amino acid sequence. Adv. Enzymol. Relat. Areas Mol. Biol. 1978, 47, 45–148. [Google Scholar] [CrossRef]
- Deleage, G.; Roux, B. An algorithm for protein secondary structure prediction based on class prediction. Protein. Eng. 1987, 1, 289–294. [Google Scholar] [CrossRef]
- Levitt, M. Conformational preferences of amino acids in globular proteins. Biochemistry 1978, 17, 4277–4285. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, Y. Protein Structure and Function Prediction Using I-TASSER. Curr. Protoc. Bioinformatics 2015, 52, 5–8. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef]
- Kuroda, K.; Caputo, G.A.; DeGrado, W.F. The role of hydrophobicity in the antimicrobial and hemolytic activities of polymethacrylate derivatives. Chemistry 2009, 15, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Sovadinova, I.; Yasuhara, K.; Vemparala, S.; Caputo, G.A.; Kuroda, K. Biomimetic antimicrobial polymers-Design, characterization, antimicrobial, and novel applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2023, 15, e1866. [Google Scholar] [CrossRef] [PubMed]
- Cuervo-Rodriguez, R.; Munoz-Bonilla, A.; Lopez-Fabal, F.; Fernandez-Garcia, M. Hemolytic and Antimicrobial Activities of a Series of Cationic Amphiphilic Copolymers Comprised of Same Centered Comonomers with Thiazole Moieties and Polyethylene Glycol Derivatives. Polymers 2020, 12, 972. [Google Scholar] [CrossRef]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of amphipathicity and hydrophobicity in the balance between hemolysis and peptide–membrane interactions of three related antimicrobial peptides. Colloids Surf. B Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Phuong, P.T.; Oliver, S.; He, J.; Wong, E.H.H.; Mathers, R.T.; Boyer, C. Effect of Hydrophobic Groups on Antimicrobial and Hemolytic Activity: Developing a Predictive Tool for Ternary Antimicrobial Polymers. Biomacromolecules 2020, 21, 5241–5255. [Google Scholar] [CrossRef] [PubMed]
- Burman, L.G.; Nordstrom, K.; Boman, H.G. Resistance of Escherichia coli to penicillins. V. Physiological comparison of two isogenic strains, one with chromosomally and one with episomally mediated ampicillin resistance. J. Bacteriol. 1968, 96, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Shirley, D.J.; Chrom, C.L.; Richards, E.A.; Carone, B.R.; Caputo, G.A. Antimicrobial activity of a porphyrin binding peptide. Pept. Sci. 2018, 110, e24074. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Length | MW | Net Charge | GRAVY a | Hydrophobicity b |
|---|---|---|---|---|---|---|
| C18G-18 | ALYKKLLKKWLKSAKKLG-NH2 | 18 | 2116.7 | +7 | −0.45 | 9.58 |
| C18G-13 | LLKKWLKSAKKLG-NH2 | 13 | 1512.9 | +5 | −0.354 | 7.24 |
| C18G-10 | KWLKSAKKLG-NH2 | 10 | 1158.5 | +4 | −0.83 | 4.97 |
| Peptide | E. coli | S. aureus | B. subtilis |
|---|---|---|---|
| C18G | 2.5 | 2.5 | 2.5 |
| C18G-18 | 5 | 2.5 | 2.5 |
| C18G-13 | 5 | >15 | 5 |
| C18G-10 | >15 | >15 | >15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meier, S.; Ridgway, Z.M.; Picciano, A.L.; Caputo, G.A. Impacts of Hydrophobic Mismatch on Antimicrobial Peptide Efficacy and Bilayer Permeabilization. Antibiotics 2023, 12, 1624. https://doi.org/10.3390/antibiotics12111624
Meier S, Ridgway ZM, Picciano AL, Caputo GA. Impacts of Hydrophobic Mismatch on Antimicrobial Peptide Efficacy and Bilayer Permeabilization. Antibiotics. 2023; 12(11):1624. https://doi.org/10.3390/antibiotics12111624
Chicago/Turabian StyleMeier, Steven, Zachary M. Ridgway, Angela L. Picciano, and Gregory A. Caputo. 2023. "Impacts of Hydrophobic Mismatch on Antimicrobial Peptide Efficacy and Bilayer Permeabilization" Antibiotics 12, no. 11: 1624. https://doi.org/10.3390/antibiotics12111624
APA StyleMeier, S., Ridgway, Z. M., Picciano, A. L., & Caputo, G. A. (2023). Impacts of Hydrophobic Mismatch on Antimicrobial Peptide Efficacy and Bilayer Permeabilization. Antibiotics, 12(11), 1624. https://doi.org/10.3390/antibiotics12111624