
Environmentally Relevant Antibiotic Concentrations Exert Stronger Selection Pressure on River Biofilm Resistomes than AMR-Reservoir Effluents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
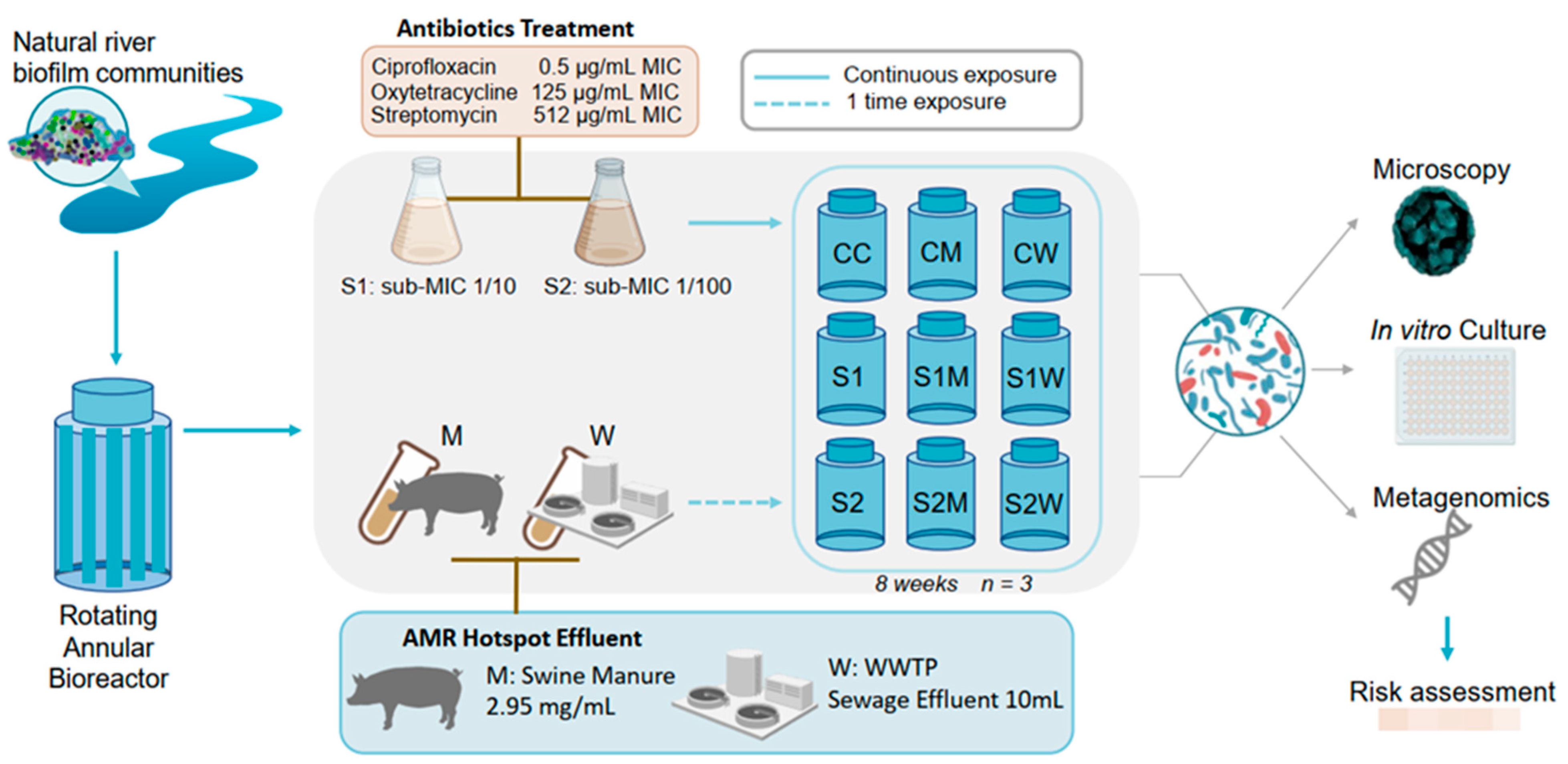
2.1. Microcosm Experimental Design: ERCA and AMR-Reservoir Treatments Exposure
2.2. Community Structure/Phenotype Using Microscopic Analysis
2.3. DNA Extraction, WSG Sequencing and Data Normalization
2.4. Metagenomic Analysis
2.5. Antimicrobial Susceptibility Testing
2.6. Statistical Analysis
3. Results and Discussion
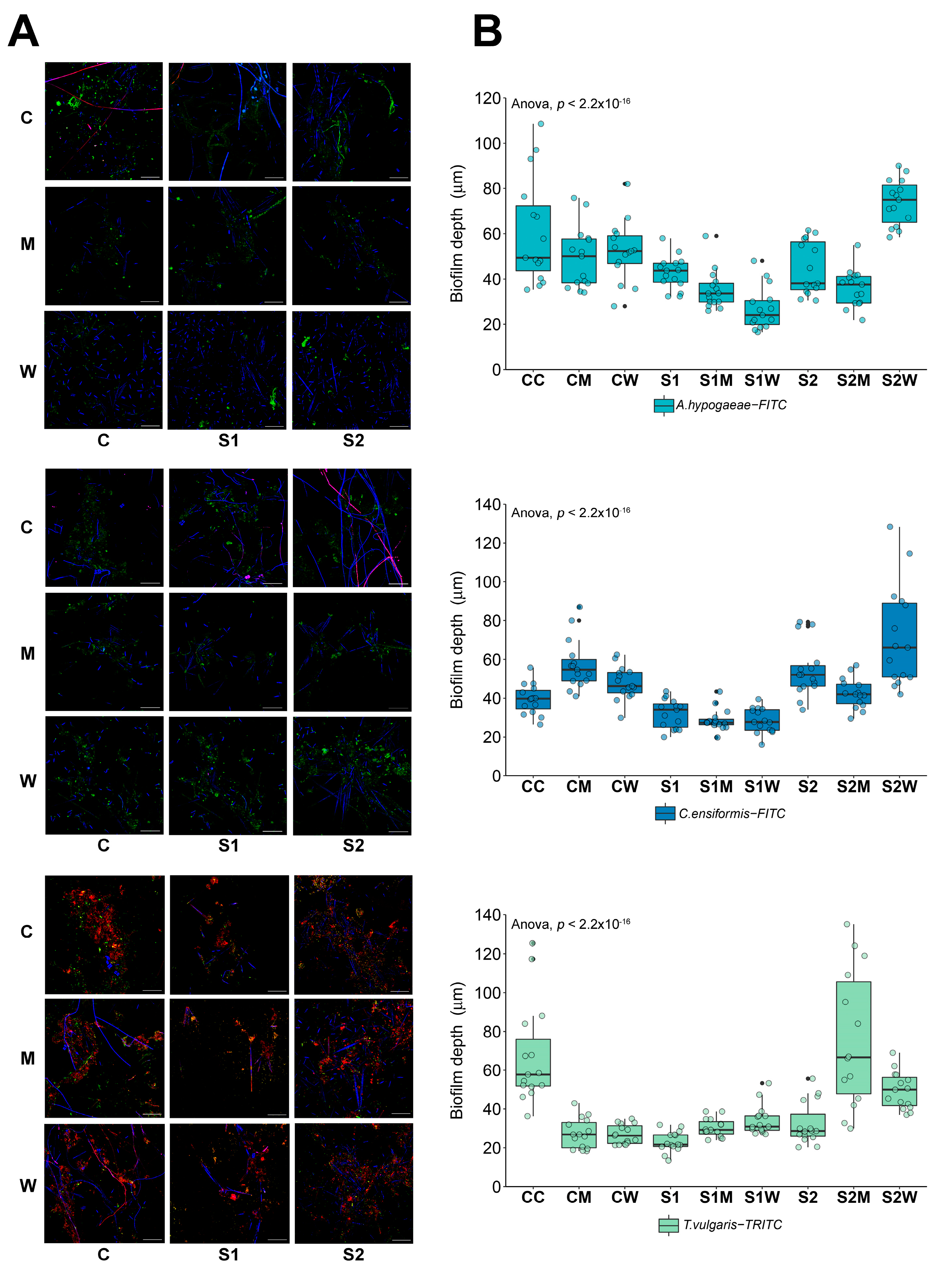
3.1. Biofilm Structure and Composition
3.2. Microbiome Composition under ERCA Exposure
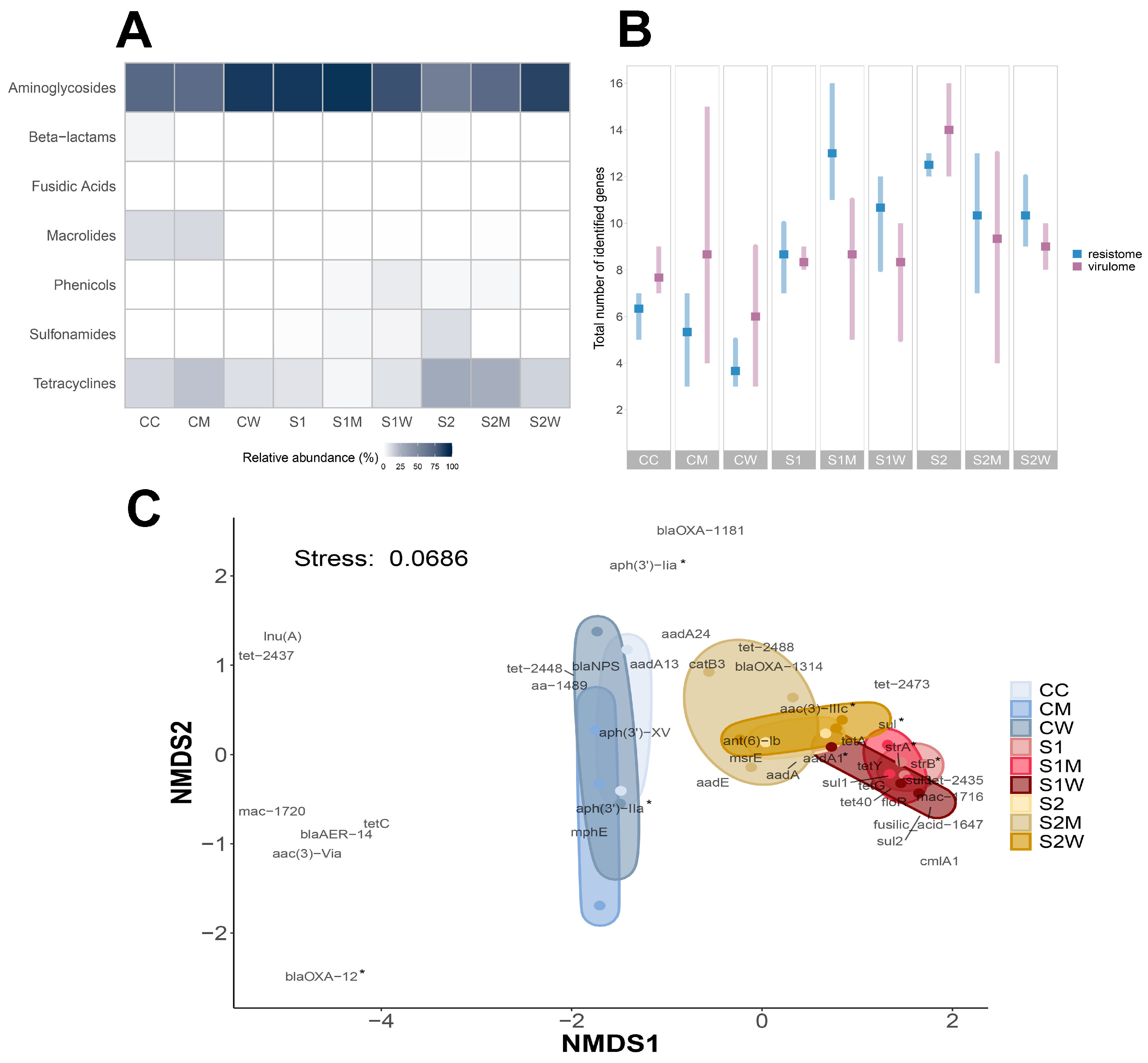
3.3. Resistome Composition at Biofilm Community Level
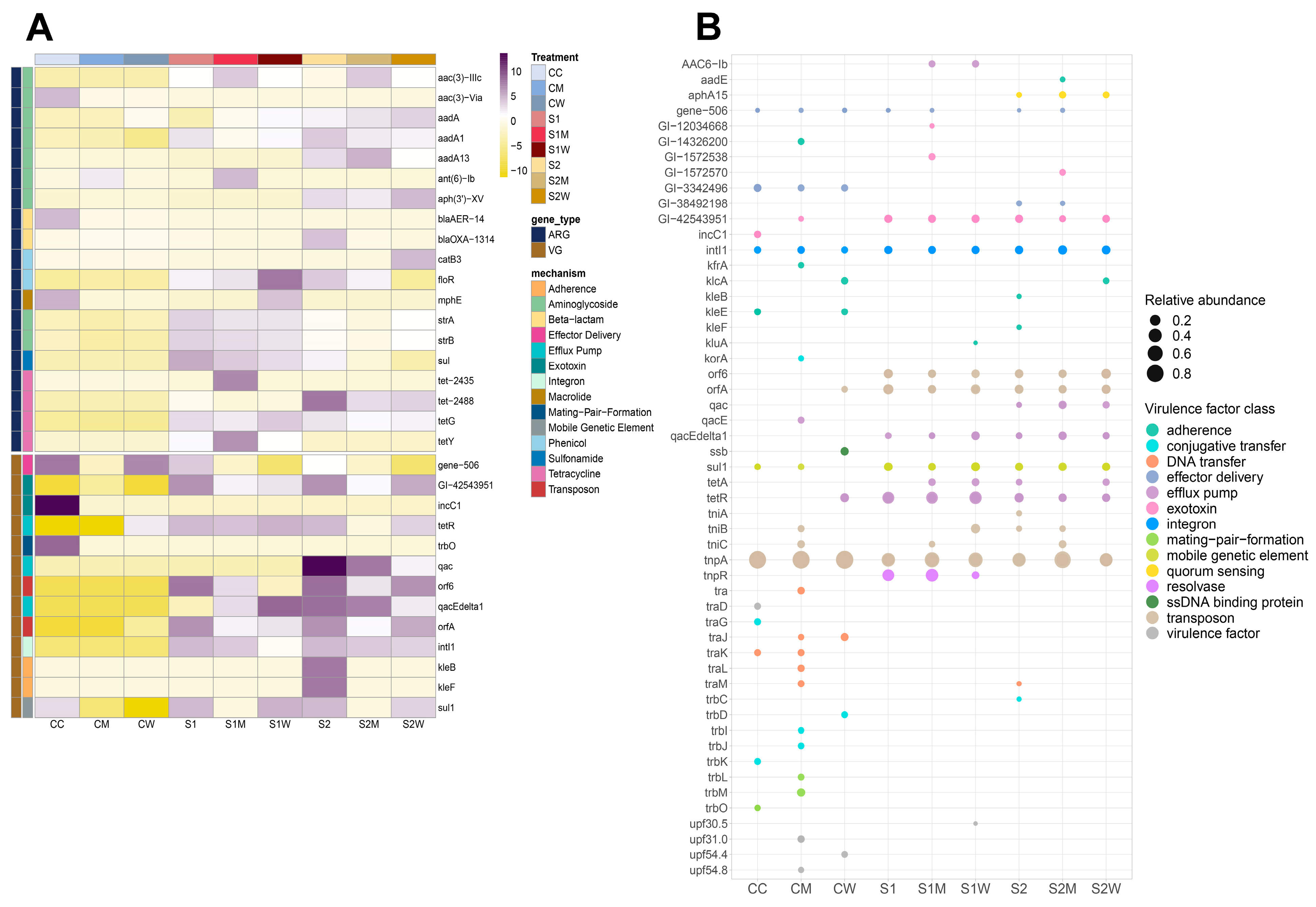
3.4. Virulome Composition at Biofilm Community Level
3.5. Mobilome Composition at Biofilm Community Level
3.6. Co-Occurrence Patterns of the Biofilm Resistome
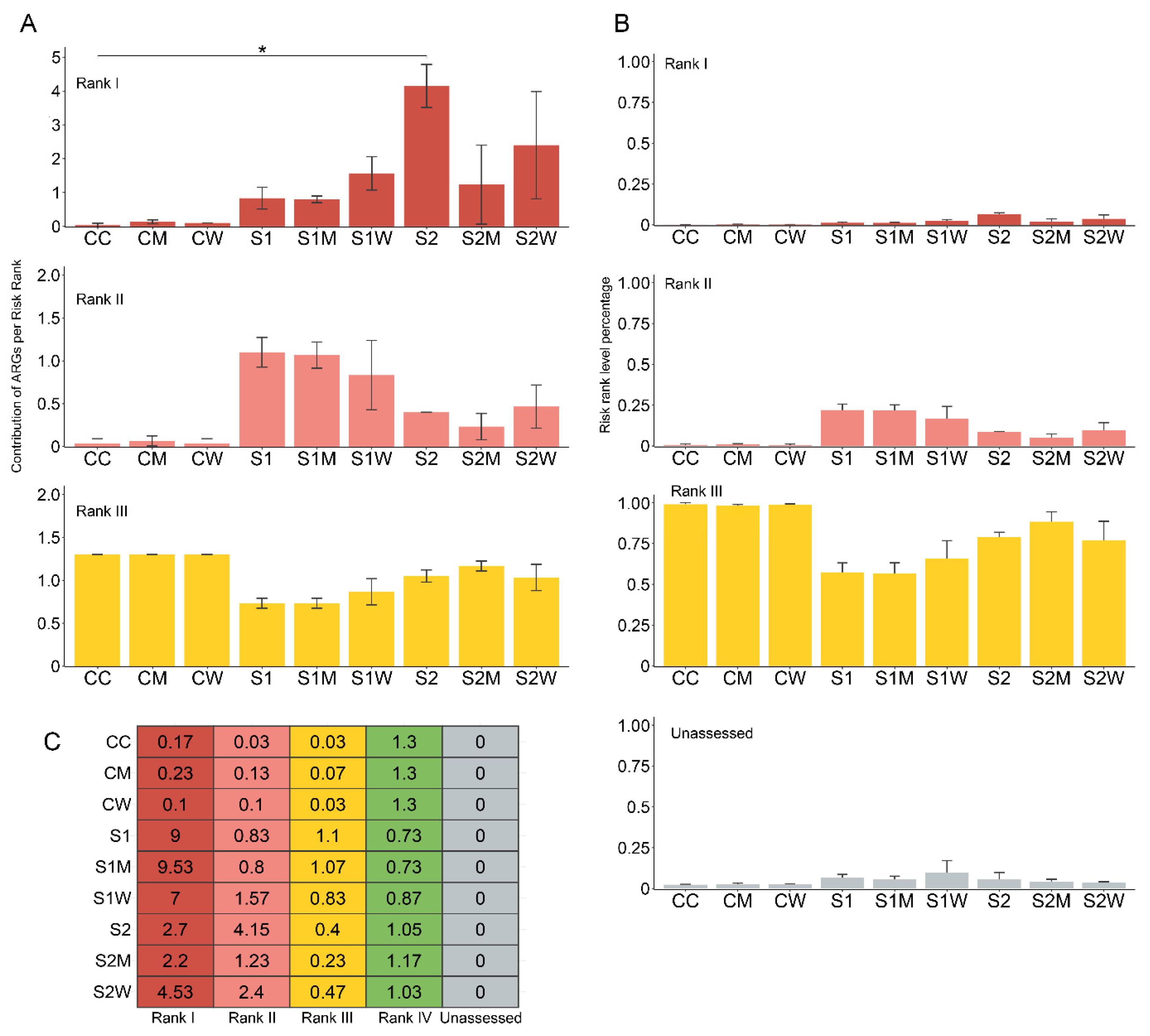
3.7. Implications to Resistome Risk Assessment in Natural Biofilms
3.8. NARMS Analysis
3.9. Predicted Community Dynamics—In Silico vs. Phenotypic Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kraemer, S.A.; Ramachandran, A.; Perron, G.G. Antibiotic Pollution in the Environment: From Microbial Ecology to Public Policy. Microorganisms 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Grenni, P. Antimicrobial Resistance in Rivers: A Review of the Genes Detected and New Challenges. Environ. Toxicol. Chem. 2022, 41, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, T.M.; Smith-Vaughan, H.; Andrews, R. Convergence of Surveillance Blind Spots with Antimicrobial Resistance Hotspots. Aust. N. Z. J. Public Health 2021, 45, 541–542. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Jin, L.; Xie, J.; Liu, H.; Zhao, J.; Ye, D.; Li, X.d. Inhalable Antibiotic Resistomes Emitted from Hospitals: Metagenomic Insights into Bacterial Hosts, Clinical Relevance, and Environmental Risks. Microbiome 2022, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.L.; Boxall, A.B.A.; Kolpin, D.W.; Leung, K.M.Y.; Lai, R.W.S.; Galbán-Malagón, C.; Adell, A.D.; Mondon, J.; Metian, M.; Marchant, R.A.; et al. Pharmaceutical Pollution of the World’s Rivers. Proc. Natl. Acad. Sci. USA 2022, 119, e2113947119. [Google Scholar] [CrossRef]
- Gullberg, E.; Cao, S.; Berg, O.G.; Ilbäck, C.; Sandegren, L.; Hughes, D.; Andersson, D.I. Selection of Resistant Bacteria at Very Low Antibiotic Concentrations. PLoS Pathog. 2011, 7, e1002158. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Larsson, D.G.J. Concentrations of Antibiotics Predicted to Select for Resistant Bacteria: Proposed Limits for Environmental Regulation. Environ. Int. 2016, 86, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.K.; Zhang, L.; Yin, X.; Zhang, T.; Buckling, A.; Snape, J.; Gaze, W.H. Novel Insights into Selection for Antibiotic Resistance in Complex Microbial Communities. mBio 2018, 9, e00969-18. [Google Scholar] [CrossRef] [PubMed]
- Maestre-Carballa, L.; Lluesma Gomez, M.; Angla Navarro, A.; Garcia-Heredia, I.; Martinez-Hernandez, F.; Martinez-Garcia, M. Insights into the Antibiotic Resistance Dissemination in a Wastewater Effluent Microbiome: Bacteria, Viruses and Vesicles Matter. Environ. Microbiol. 2019, 21, 4582–4596. [Google Scholar] [CrossRef]
- Chow, L.K.M.; Ghaly, T.M.; Gillings, M.R. A Survey of Sub-Inhibitory Concentrations of Antibiotics in the Environment. J. Environ. Sci. 2021, 99, 21–27. [Google Scholar] [CrossRef]
- Marti, E.; Variatza, E.; Balcazar, J.L. The Role of Aquatic Ecosystems as Reservoirs of Antibiotic Resistance. Trends Microbiol. 2014, 22, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.; Waldron, L.; Gillings, M.R. Potential Impacts of Aquatic Pollutants: Sub-Clinical Antibiotic Concentrations Induce Genome Changes and Promote Antibiotic Resistance. Front. Microbiol. 2015, 6, 803. [Google Scholar] [CrossRef] [PubMed]
- Flores-Vargas, G.; Korber, D.R.; Bergsveinson, J. Sub-MIC Antibiotics Influence the Microbiome, Resistome and Structure of Riverine Biofilm Communities. Front. Microbiol. 2023, 14, 1194952. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Q.E.; Zhou, X.; Wang, F.H.; Muurinen, J.; Virta, M.P.; Brandt, K.K.; Zhu, Y.G. Antibiotic Resistome in the Livestock and Aquaculture Industries: Status and Solutions. Crit. Rev. Environ. Sci. Technol. 2021, 51, 2159–2196. [Google Scholar] [CrossRef]
- Gao, F.Z.; He, L.Y.; Liu, Y.S.; Zhao, J.L.; Zhang, T.; Ying, G.G. Integrating Global Microbiome Data into Antibiotic Resistance Assessment in Large Rivers. Water Res. 2024, 250, 121030. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Yang, J.; Kim, S.; Park, J.; Kim, M.; Park, W. Occurrence of Antibiotic Resistance Genes and Multidrug-Resistant Bacteria during Wastewater Treatment Processes. Sci. Total Environ. 2022, 811, 152331. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gong, Z.; Wang, S.; Song, L. Occurrence and Prevalence of Antibiotic Resistance Genes and Pathogens in an Industrial Park Wastewater Treatment Plant. Sci. Total Environ. 2023, 880, 163278. [Google Scholar] [CrossRef]
- Hutinel, M.; Fick, J.; Larsson, D.G.J.; Flach, C.F. Investigating the Effects of Municipal and Hospital Wastewaters on Horizontal Gene Transfer. Environ. Pollut. 2021, 276, 116733. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C.; van Hullebusch, E.D.; Neu, T.R.; Nielsen, P.H.; Seviour, T.; Stoodley, P.; Wingender, J.; Wuertz, S. The Biofilm Matrix: Multitasking in a Shared Space. Nat. Rev. Microbiol. 2022, 21, 70–86. [Google Scholar] [CrossRef]
- Balcázar, J.L.; Subirats, J.; Borrego, C.M. The Role of Biofilms as Environmental Reservoirs of Antibiotic Resistance. Front. Microbiol. 2015, 6, 1216. [Google Scholar] [CrossRef]
- Guo, X.P.; Yang, Y.; Lu, D.P.; Niu, Z.S.; Feng, J.N.; Chen, Y.R.; Tou, F.Y.; Garner, E.; Xu, J.; Liu, M.; et al. Biofilms as a Sink for Antibiotic Resistance Genes (ARGs) in the Yangtze Estuary. Water Res. 2018, 129, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Nomura, N.; Suzuki, S. Biofilms: Hot Spots of Horizontal Gene Transfer (HGT) in Aquatic Environments, with a Focus on a New HGT Mechanism. FEMS Microbiol. Ecol. 2020, 96, fiaa031. [Google Scholar] [CrossRef] [PubMed]
- Flores-Vargas, G.; Bergsveinson, J.; Lawrence, J.R.; Korber, D.R. Environmental Biofilms as Reservoirs for Antimicrobial Resistance. Front. Microbiol. 2021, 12, 766242. [Google Scholar] [CrossRef] [PubMed]
- Matviichuk, O.; Mondamert, L.; Geffroy, C.; Gaschet, M.; Dagot, C.; Labanowski, J. River Biofilms Microbiome and Resistome Responses to Wastewater Treatment Plant Effluents Containing Antibiotics. Front. Microbiol. 2022, 13, 795206. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, E.L.; Nietch, C.T.; Blocksom, K.; Lazorchak, J.M.; Batt, A.L.; Griffiths, R.; Klemm, D.J. Temporal Dynamics of Periphyton Exposed to Tetracycline in Stream Mesocosms. Environ. Sci. Technol. 2011, 45, 10684–10690. [Google Scholar] [CrossRef] [PubMed]
- White, A.; Hughes, J.M. Critical Importance of a One Health Approach to Antimicrobial Resistance. Ecohealth 2019, 16, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, J.M.; Fu, Y.; Mei, Z.; Schäffer, A.; Dou, Q.; Amelung, W.; Elsner, M.; Adu-Gyamfi, J.; Heng, L.; Virta, M.; et al. Antibiotic Resistance Genes in Food Production Systems Support One Health Opinions. Curr. Opin. Environ. Sci. Health 2023, 34, 100492. [Google Scholar] [CrossRef]
- Lawrence, J.R.; Swerhone, G.D.W.; Topp, E.; Korber, D.R.; Neu, T.R.; Wassenaar, L.I. Structural and Functional Responses of River Biofilm Communities to the Nonsteroidal Anti-Inflammatory Diclofenac. Environ. Toxicol. Chem. 2007, 26, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.R.; Zhu, B.; Swerhone, G.D.W.; Roy, J.; Wassenaar, L.I.; Topp, E.; Korber, D.R. Comparative Microscale Analysis of the Effects of Triclosan and Triclocarban on the Structure and Function of River Biofilm Communities. Sci. Total Environ. 2009, 407, 3307–3316. [Google Scholar] [CrossRef]
- WHO. Integrated Surveillance of Antimicrobial Resistance in Foodborne Bacteria: Application of a One Health Approach; WHO: Geneva, Switzerland, 2017; ISBN 978-92-4-151241-1. [Google Scholar]
- Chekabab, S.M.; Lawrence, J.R.; Alvarado, A.; Predicala, B.; Korber, D.R. A Health Metadata-Based Management Approach for Comparative Analysis of High-Throughput Genetic Sequences for Quantifying Antimicrobial Resistance Reduction in Canadian Hog Barns. Comput. Struct. Biotechnol. J. 2020, 18, 2629–2638. [Google Scholar] [CrossRef]
- Barrios, R.E.; Khuntia, H.K.; Bartelt-Hunt, S.L.; Gilley, J.E.; Schmidt, A.M.; Snow, D.D.; Li, X. Fate and Transport of Antibiotics and Antibiotic Resistance Genes in Runoff and Soil as Affected by the Timing of Swine Manure Slurry Application. Sci. Total Environ. 2020, 712, 136505. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.C.; Duerschner, J.; Gilley, J.E.; Schmidt, A.M.; Bartelt-Hunt, S.L.; Snow, D.D.; Eskridge, K.M.; Li, X. Antibiotic Resistance Genes in Swine Manure Slurry as Affected by Pit Additives and Facility Disinfectants. Sci. Total Environ. 2021, 761, 143287. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, X.; Zheng, S.; Kai, Z.; Jin, T.; Shi, R.; Huang, H.; Zheng, X. Effects of Wastewater Treatment and Manure Application on the Dissemination of Antimicrobial Resistance around Swine Feedlots. J. Clean. Prod. 2021, 280, 123794. [Google Scholar] [CrossRef]
- Dynes, J.J.; Lawrence, J.R.; Korber, D.R.; Swerhone, G.D.W.; Leppard, G.G.; Hitchcock, A.P. Quantitative Mapping of Chlorhexidine in Natural River Biofilms. Sci. Total Environ. 2006, 369, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Neu, T.R.; Swerhone, G.D.W.; Lawrence, J.R. Assessment of Lectin-Binding Analysis for in Situ Detection of Glycoconjugates in Biofilm Systems. Microbiology 2001, 147, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.R.; Swerhone, G.D.W.; Wassenaar, L.I.; Neu, T.R. Effects of Selected Pharmaceuticals on Riverine Biofilm Communities. Can. J. Microbiol. 2005, 51, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Bergsveinson, J.; Roy, J.; Maynard, C.; Sanschagrin, S.; Freeman, C.N.; Swerhone, G.D.W.; Dynes, J.J.; Tremblay, J.; Greer, C.W.; Korber, D.R.; et al. Metatranscriptomic Insights Into the Response of River Biofilm Communities to Ionic and Nano-Zinc Oxide Exposures. Front. Microbiol. 2020, 11, 267. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Connelly, S.; Bristol, J.A.; Hubert, S.; Subramanian, P.; Hasan, N.A.; Colwell, R.R.; Kaleko, M. SYN-004 (Ribaxamase), an Oral Beta-Lactamase, Mitigates Antibiotic-Mediated Dysbiosis in a Porcine Gut Microbiome Model. J. Appl. Microbiol. 2017, 123, 66–79. [Google Scholar] [CrossRef]
- Yan, Q.; Wi, Y.M.; Thoendel, M.J.; Raval, Y.S.; Greenwood-Quaintance, K.E.; Abdel, M.P.; Jeraldo, P.R.; Chia, N.; Patel, R. Evaluation of the CosmosID Bioinformatics Platform for Prosthetic Joint-Associated Sonicate Fluid Shotgun Metagenomic Data Analysis. J. Clin. Microbiol. 2019, 57, e01182-18. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, A.C.; Ratan, A.; Acerbi, E.; Drautz-Moses, D.I.; Premkrishnan, B.N.V.; Costea, P.I.; Linz, B.; Purbojati, R.W.; Paulo, D.F.; Gaultier, N.E.; et al. The Microbiomes of Blowflies and Houseflies as Bacterial Transmission Reservoirs. Sci. Rep. 2017, 7, 16324. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2016, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Abricate. Available online: https://github.com/tseemann/abricate (accessed on 28 February 2022).
- Zhang, A.N.; Gaston, J.M.; Dai, C.L.; Zhao, S.; Poyet, M.; Groussin, M.; Yin, X.; Li, L.G.; van Loosdrecht, M.C.M.; Topp, E.; et al. An Omics-Based Framework for Assessing the Health Risk of Antimicrobial Resistance Genes. Nat. Commun. 2021, 12, 4765. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation: Vienna, Austria, 2020. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.B.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package 2020, R Package Version 2.5-7. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 30 April 2022).
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize Implements and Enhances Circular Visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. Proc. Int. AAAI Conf. Web Soc. Media 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Love, M.A.; Ahlmann-eltze, C.; Forbes, K.; Anders, S.; Huber, W. Package ‘DESeq2’. 2022. Available online: https://bioconductor.org/packages/release/bioc/html/DESeq2.html (accessed on 30 April 2022).
- Yang, C.; Mai, J.; Cao, X.; Burberry, A.; Cominelli, F.; Zhang, L. Ggpicrust2: An R Package for PICRUSt2 Predicted Functional Profile Analysis and Visualization. Bioinformatics 2023, 39, btad470. [Google Scholar] [CrossRef]
- Cerca, N.; Martins, S.; Sillankorva, S.; Jefferson, K.K.; Pier, G.B.; Oliveira, R.; Azeredo, J. Effects of Growth in the Presence of Subinhibitory Concentrations of Dicloxacillin on Staphylococcus Epidermidis and Staphylococcus Haemolyticus Biofilms. Appl. Environ. Microbiol. 2005, 71, 8677–8682. [Google Scholar] [CrossRef]
- Seke Etet, P.F.; Nwabo Kamdje, A.H.; Vecchio, L.; Farahna, M.; Mahomoodally, M.F. Signaling Pathways Sustaining Antibiotic Resistance: Lessons from Methicillin-Resistant Staphylococcus Aureus. Lessons from Methicillin-Resistant Staphylococcus Aureus; Elsevier Inc.: Amsterdam, The Netherlands, 2016; ISBN 9780128036686. [Google Scholar]
- Qin, Z.; Zhao, Z.; Xia, L.; Ohore, O.E. Research Trends and Hotspots of Aquatic Biofilms in Freshwater Environment during the Last Three Decades: A Critical Review and Bibliometric Analysis. Environ. Sci. Pollut. Res. 2022, 29, 47915–47930. [Google Scholar] [CrossRef] [PubMed]
- Bruchmann, J.; Kirchen, S.; Schwartz, T. Sub-Inhibitory Concentrations of Antibiotics and Wastewater Influencing Biofilm Formation and Gene Expression of Multi-Resistant Pseudomonas Aeruginosa Wastewater Isolates. Environ. Sci. Pollut. Res. 2013, 20, 3539–3549. [Google Scholar] [CrossRef] [PubMed]
- Erken, M.; Lutz, C.; McDougald, D. The Rise of Pathogens: Predation as a Factor Driving the Evolution of Human Pathogens in the Environment. Microb. Ecol. 2013, 65, 860–868. [Google Scholar] [CrossRef] [PubMed]
- Świacka, K.; Maculewicz, J.; Kowalska, D.; Grace, M.R. Do Pharmaceuticals Affect Microbial Communities in Aquatic Environments? A Review. Front. Environ. Sci. 2023, 10, 1093920. [Google Scholar] [CrossRef]
- Lawrence, J.R.; Paule, A.; Swerhone, G.D.W.; Roy, J.; Grigoryan, A.A.; Dynes, J.J.; Chekabab, S.M.; Korber, D.R. Microscale and Molecular Analyses of River Biofilm Communities Treated with Microgram Levels of Cerium Oxide Nanoparticles Indicate Limited but Significant Effects. Environ. Pollut. 2020, 256, 113515. [Google Scholar] [CrossRef] [PubMed]
- Brumfield, K.D.; Hasan, N.A.; Leddy, M.B.; Cotruvo, J.A.; Rashed, S.M.; Colwell, R.R.; Huq, A. A Comparative Analysis of Drinking Water Employing Metagenomics. PLoS ONE 2020, 15, e0231210. [Google Scholar] [CrossRef]
- Guo, X.p.; Sun, X.; Chen, Y.; Hou, L.; Liu, M.; Yang, Y. Antibiotic Resistance Genes in Biofilms on Plastic Wastes in an Estuarine Environment. Sci. Total Environ. 2020, 745, 140916. [Google Scholar] [CrossRef] [PubMed]
- Decewicz, P.; Kitowicz, M.; Radlinska, M. Characteristics and Comparative Genomic Analysis of a Novel Virus, VarioGold, the First Bacteriophage of Variovorax. Int. J. Mol. Sci. 2022, 23, 13539. [Google Scholar] [CrossRef]
- Wingender, J.; Flemming, H.C. Biofilms in Drinking Water and Their Role as Reservoir for Pathogens. Int. J. Hyg. Environ. Health 2011, 214, 417–423. [Google Scholar] [CrossRef]
- Tarek, M.H.; Garner, E. A Proposed Framework for the Identification of Indicator Genes for Monitoring Antibiotic Resistance in Wastewater: Insights from Metagenomic Sequencing. Sci. Total Environ. 2023, 854, 158698. [Google Scholar] [CrossRef]
- Mao, G.; Liang, J.; Wang, Q.; Zhao, C.; Bai, Y.; Liu, R.; Liu, H.; Qu, J. Epilithic Biofilm as a Reservoir for Functional Virulence Factors in Wastewater-Dominant Rivers after WWTP Upgrade. J. Environ. Sci. 2021, 101, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gu, A.Z.; Cen, T.; Li, X.; He, M.; Li, D.; Chen, J. Sub-Inhibitory Concentrations of Heavy Metals Facilitate the Horizontal Transfer of Plasmid-Mediated Antibiotic Resistance Genes in Water Environment. Environ. Pollut. 2018, 237, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.; Organiscak, M.; Dieter, L.; Shingleton, C.; Haddix, M.; Joshi, S.; Pruden, A.; Ashbolt, N.J.; Medema, G.; Hamilton, K.A. Towards Risk Assessment for Antibiotic Resistant Pathogens in Recycled Water: A Systematic Review and Summary of Research Needs. Environ. Microbiol. 2021, 23, 7355–7372. [Google Scholar] [CrossRef] [PubMed]
- Liguori, K.; Calarco, J.; Rivera, G.M.; Kurowski, A.; Keenum, I.; Davis, B.C.; Harwood, V.J.; Pruden, A. Comparison of Cefotaxime-Resistant Escherichia coli and sul1 and intI1 by qPCR for Monitoring of Antibiotic Resistance of Wastewater, Surface Water, and Recycled Water. Antibiotics 2023, 12, 1252. [Google Scholar] [CrossRef] [PubMed]
- Risely, A.; Newbury, A.; Stalder, T.; Simmons, B.I.; Top, E.M.; Buckling, A.; Sanders, D. Host- Plasmid Network Structure in Wastewater Is Linked to Antimicrobial Resistance Genes. Nat. Commun. 2024, 15, 555. [Google Scholar] [CrossRef] [PubMed]
- Emond-Rheault, J.G.; Hamel, J.; Jeukens, J.; Freschi, L.; Kukavica-Ibrulj, I.; Boyle, B.; Tamber, S.; Malo, D.; Franz, E.; Burnett, E.; et al. The Salmonella Enterica Plasmidome as a Reservoir of Antibiotic Resistance. Microorganisms 2020, 8, 1016. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, D.E.; Tietze, E. Comparative Biology of IncQ and IncQ-Like Plasmids. Microbiol. Mol. Biol. Rev. 2001, 65, 481–496. [Google Scholar] [CrossRef] [PubMed]
- McMillan, E.A.; Jackson, C.R.; Frye, J.G. Transferable Plasmids of Salmonella Enterica Associated With Antibiotic Resistance Genes. Front. Microbiol. 2020, 11, 562181. [Google Scholar] [CrossRef]
- Arredondo-Alonso, S.; Willems, R.J.; van Schaik, W.; Schürch, A.C. On the (Im)Possibility of Reconstructing Plasmids from Whole-Genome Short-Read Sequencing Data. Microb. Genomics 2017, 3, e000128. [Google Scholar] [CrossRef]
- Guliy, O.I.; Evstigneeva, S.S.; Karavaeva, O.A. Antimicrobial Resistance and Current Methods for Its Detection. Front. Biosci. Elit. 2023, 15, 19. [Google Scholar] [CrossRef]
- Matviichuk, O.; Mondamert, L.; Geffroy, C.; Dagot, C.; Labanowski, J. Life in an Unsuspected Antibiotics World: River Biofilms. Water Res. 2023, 231, 119611. [Google Scholar] [CrossRef] [PubMed]
- Subirats, J.; Sharpe, H.; Tai, V.; Fruci, M.; Topp, E. Metagenome Meta-Analysis Reveals an Increase in the Abundance of Some Multidrug Efflux Pumps and Mobile Genetic Elements in Chemically Polluted Environments. Appl. Environ. Microbiol. 2023, 89, e01047-23. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.K.; Stanton, I.; Gaze, W.H.; Snape, J. Dawning of a New ERA: Environmental Risk Assessment of Antibiotics and Their Potential to Select for Antimicrobial Resistance. Water Res. 2021, 200, 117233. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Tagaki, C.; Fukuda, A.; Okubo, T.; Boonla, C.; Suzuki, S.; Seki, K.; Takada, H.; Tamura, Y. Use of Aeromonas Spp. as General Indicators of Antimicrobial Susceptibility among Bacteria in Aquatic Environments in Thailand. Front. Microbiol. 2016, 7, 710. [Google Scholar] [CrossRef] [PubMed]
- Liguori, K.; Keenum, I.; Davis, B.C.; Calarco, J.; Milligan, E.; Harwood, V.J.; Pruden, A. Antimicrobial Resistance Monitoring of Water Environments: A Framework for Standardized Methods and Quality Control. Environ. Sci. Technol. 2022, 56, 9149–9160. [Google Scholar] [CrossRef] [PubMed]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating Taxonomic, Functional, and Strain-Level Profiling of Diverse Microbial Communities with BioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, Information, Knowledge and Principle: Back to Metabolism in KEGG. Nucleic Acids Res. 2013, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Wu, J.; Hu, Y.; Sha, C.; Yao, S.; Li, P.; Lin, K.; Cui, C. Occurrence of Heavy Metals, Antibiotics, and Antibiotic Resistance Genes in Different Kinds of Land-Applied Manure in China. Environ. Sci. Pollut. Res. 2021, 28, 40011–40021. [Google Scholar] [CrossRef]
- Bharti, R.; Grimm, D.G. Current Challenges and Best-Practice Protocols for Microbiome Analysis. Brief. Bioinform. 2021, 22, 178–193. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.-F. Molecular Mechanisms of Biofilm-Based Antibiotic Resistance and Tolerance in Pathogenic Bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, Y.S.; Zhao, J.L.; Liu, W.R.; He, L.Y.; Zhang, J.N.; Chen, J.; He, L.K.; Zhang, Q.Q.; Ying, G.G. Occurrence, Fate and Mass Loadings of Antibiotics in Two Swine Wastewater Treatment Systems. Sci. Total Environ. 2018, 639, 1421–1431. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Vargas, G.; Bergsveinson, J.; Korber, D.R. Environmentally Relevant Antibiotic Concentrations Exert Stronger Selection Pressure on River Biofilm Resistomes than AMR-Reservoir Effluents. Antibiotics 2024, 13, 539. https://doi.org/10.3390/antibiotics13060539
Flores-Vargas G, Bergsveinson J, Korber DR. Environmentally Relevant Antibiotic Concentrations Exert Stronger Selection Pressure on River Biofilm Resistomes than AMR-Reservoir Effluents. Antibiotics. 2024; 13(6):539. https://doi.org/10.3390/antibiotics13060539
Chicago/Turabian StyleFlores-Vargas, Gabriela, Jordyn Bergsveinson, and Darren R. Korber. 2024. "Environmentally Relevant Antibiotic Concentrations Exert Stronger Selection Pressure on River Biofilm Resistomes than AMR-Reservoir Effluents" Antibiotics 13, no. 6: 539. https://doi.org/10.3390/antibiotics13060539
APA StyleFlores-Vargas, G., Bergsveinson, J., & Korber, D. R. (2024). Environmentally Relevant Antibiotic Concentrations Exert Stronger Selection Pressure on River Biofilm Resistomes than AMR-Reservoir Effluents. Antibiotics, 13(6), 539. https://doi.org/10.3390/antibiotics13060539