

Acquired Bacterial Resistance to Antibiotics and Resistance Genes: From Past to Future

 , , , , , ,
, , , , , ,  ,
,  ,
,  and
and
Abstract
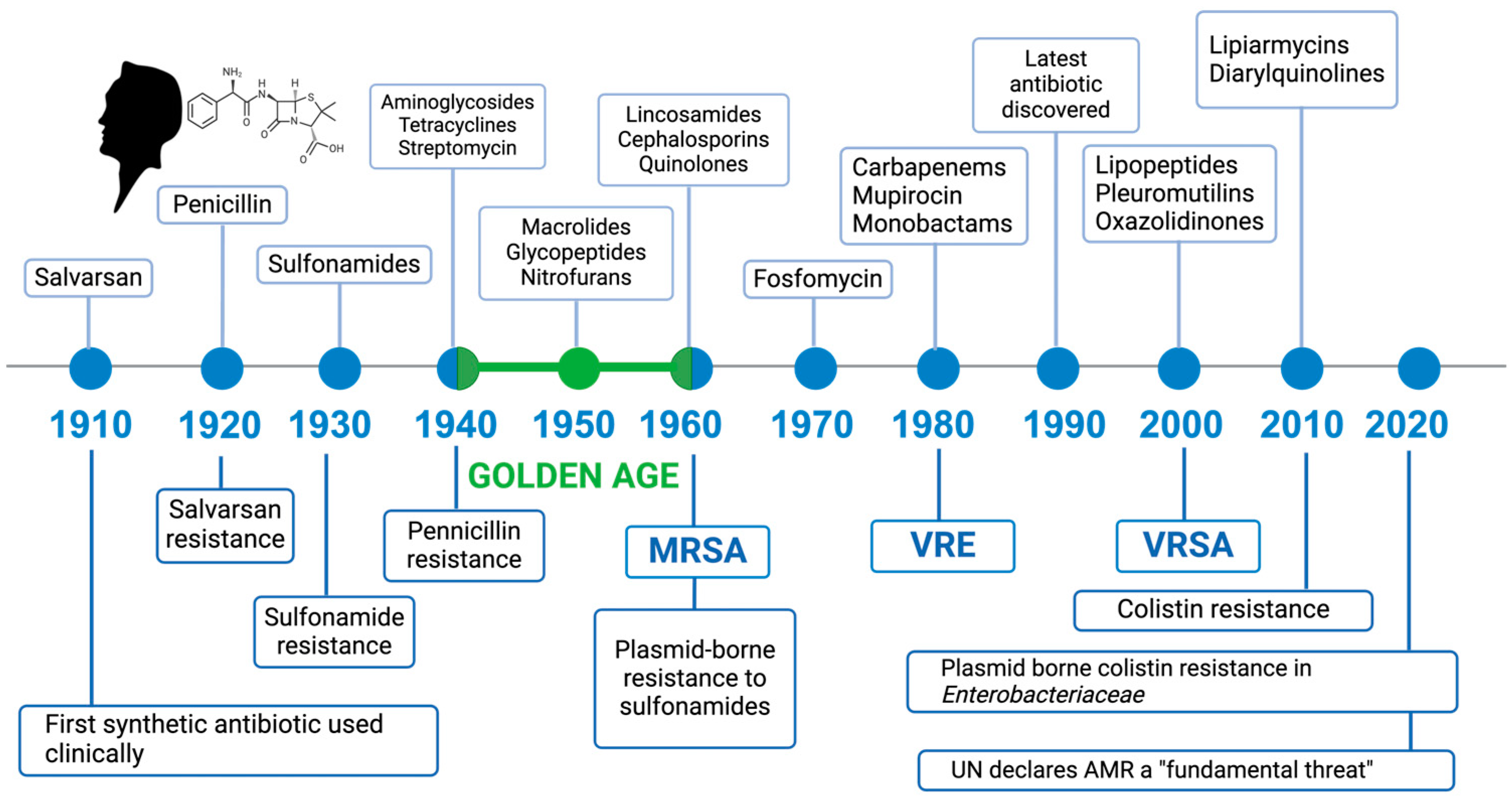
:1. Introduction
2. Resistance or Persistence?
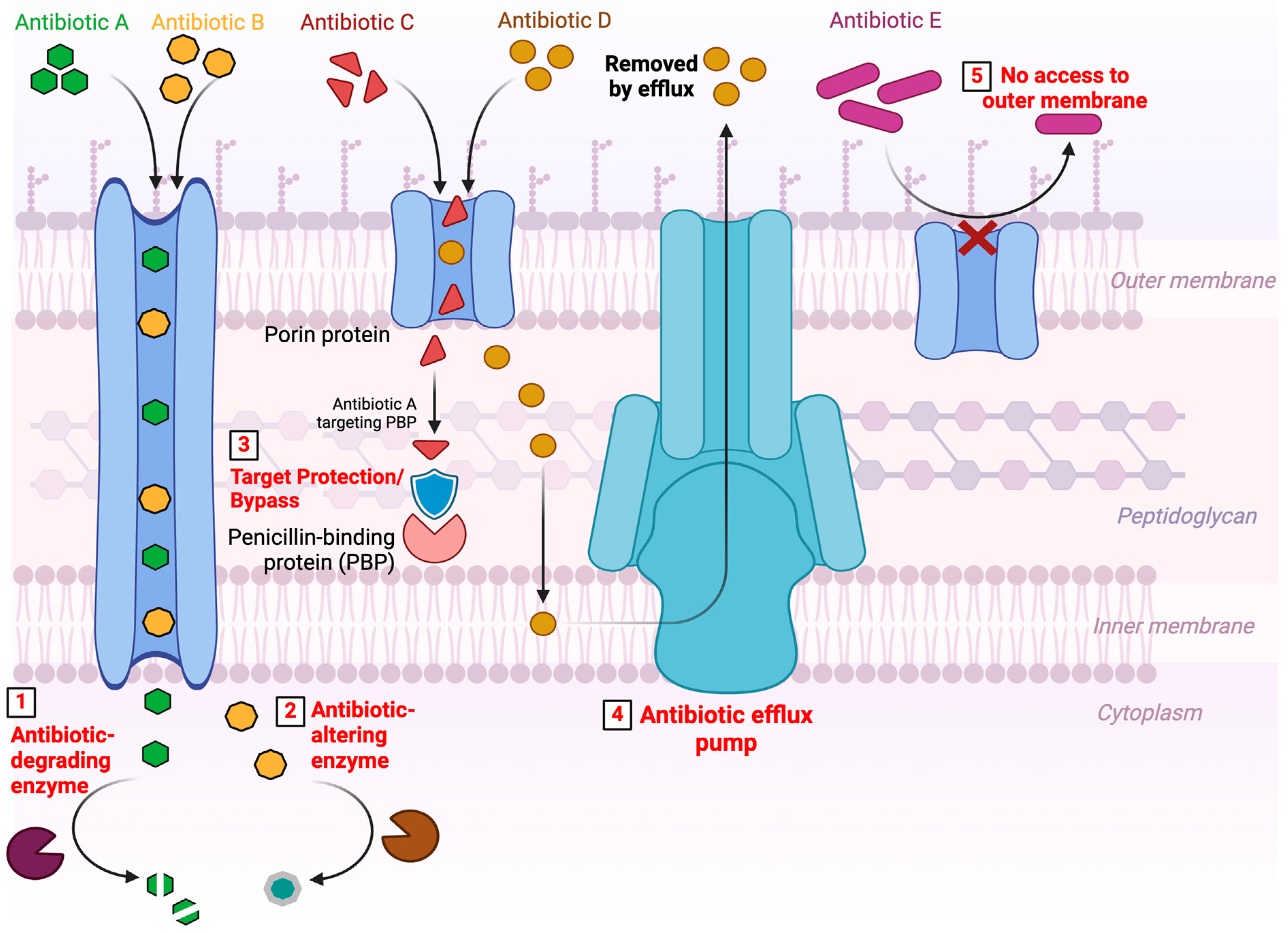
3. Antimicrobial Resistance Mechanisms
4. Factors Affecting the Acquisition of Resistance Genes
- (i)
- Host-encoded specific factors: There are several systems with which bacteria protect themselves from exogenous DNA. The most common are restriction/editing systems and CRISPR-Cas systems (acronym for regularly interspaced short palindromic repeats/CRISPR-associated protein), immuno-adaptive defense mechanisms used by archaea and bacteria capable of identifying and degrading incoming foreign genetic material [63,64]. Both systems can reduce the spread of phage DNA, integrative conjugative elements (ICEs), and plasmids.
- (ii)
- Non-specific host factors: In this case, bacteria do not possess the species-specific target site for a given integrative conjugative element (ICE), or host replication systems prevent plasmid replication. In addition to endogenous systems, cell surface architecture may also hinder conjugation by reducing the productive functionality of mobile genetic element transfer. Furthermore, Gram-positive and Gram-negative bacteria produce a wide range of inhibitory substances and antimicrobial products to protect themselves from the constant assault of bacteriophages, the most common of which are the colicin bacteriocins produced by E. coli [65].
- (iii)
- Genetic element-encoded factors: To overcome bacterial defense systems, mobile genetic elements such as plasmids and ICEs can encode anti-restriction proteins that inactivate the host’s restriction system, allowing the MGE to enter the new host without being degraded. In addition, some genes encode anti-restriction proteins that mimic the structure of DNA, exhibiting DNA-like negative surface charge distributions, which are recognized and bound by the restriction enzyme [66].
- (iv)
- Environmental factors: The gene transfer for the spread of resistance is influenced by the presence of antibiotics in the environment and is favored in environments with relatively high density, such as the intestine and oral cavity, or in biofilm.
5. Mechanism of Resistance
- (i)
- Structural modifications of the antimicrobial molecule
- (a)
- The production of enzymes capable of introducing chemical changes in the anti-microbial molecular structure is a mechanism of acquired resistance long known in both Gram-negative and Gram-positive bacteria. There are several modifying enzymes that catalyze the reactions through acetylation (AAC, Aminoglycoside N-acetyltransferase; CAT, Chloramphenicol acetyltransferase; VAT, virginiamycin O-acetyltransferase), phosphorylation (APH, Aminoglycoside phosphotransferase; CPT, Chloramphenicol O-phosphotransferase), and adenylation (ANT, Aminoglycosides adenylyltransferase, LIN Lincosamide adenylyltransferase). These enzymes belong to the transferase family, a large superfamily of enzymes that differ in terms of substrate specificity and mechanism of action, capable to covalently bind various chemical groups [67,71,72]. Regardless of the biochemical reaction, the resulting effect is often related to a steric disorder that decreases the drug’s avidity towards its target [73].
- (b)
- The mechanism of destruction of antibiotic molecules involves the inactivation of the antibiotic’s active ingredient through their degradation. β-lactamases and macrolide esterases that destroy β-lactams and macrolides, respectively, are the most common enzymes that catalyze antibiotic hydrolysis. β-lactamases, first described in the early 1940s, belong to a superfamily of enzymes that currently has more than 2000 members [92]. They are responsible for the hydrolysis of the amide bond in the β-lactam ring, the common structural element of all β-lactam antibiotics (penicillins, cephalosporins, carbapenems, and monobactams) [93]. In an attempt to group this large number of enzymes, two main, not entirely overlapping, classification schemes have been proposed: (1) the Ambler classification, based on amino acid sequence identity and separating the β-lactamases into four groups, in which the enzymes of classes A (CTX-M, Cefotaximase; TEM, Temoniera of the patient in which it was originally found; SHV, variable sulfhydryl reagent; and KPC, Klebsiella pneumoniae carbapenemase), C (BLC, C β-lactamases, also known as AmpC or cephalosporinase), and D (OXA, Oxacillinase) are classified as serine hydrolases, while the enzymes of class B (NDM, New Delhi metallo-β-lactamase; VIM, metallo-β-lactamase encoded by the Verona integron; and IMP, imipenemase) are metalloenzymes; (2) the Bush-Jacoby classification, which divides β-lactamases into four categories (each with different subgroups) based on their biochemical function, mainly based on substrate specificity [94,95]. The high mutation rate of β-lactamases contributes to the rapid spread of resistant bacteria [94]. Also noteworthy and posing a global threat is the detection in hospital clinical samples of pathogenic bacteria carrying up to eight β-lactamase genes simultaneously, which makes them capable of hydrolyzing most penicillins and cephalosporins, adversely affecting clinical and therapeutic outcomes, with higher rates of morbidity and mortality, longer hospital stays and high healthcare costs [96]. In 2004, a strain of Klebsiella pneumonia was described, which was isolated from seven New York City hospitals and produced up to 10 different β-lactamases, including a FOX-like plasmid-mediated AmpC, in addition to the previously reported KPC, SHV ESBL, and IRT β-lactamases [97].
- (ii)
- Antibiotic target site protection
- (a)
- As mentioned, there is an innate resistance in the ability of bacteria to limit the entry of antimicrobial agents into the cell. This natural difference is given by the cell wall, which in Gram-negative bacteria is quite complex, and provides a barrier to certain types of antimicrobial molecules [70]. In fact, it prevents the penetration of hydrophilic drugs, such as β-lactams, tetracyclines, and some fluoroquinolones and, therefore, their binding to the target site. In these bacteria, hydrophilic molecules are endocytosed through diffusion channels and porins [105,106]. However, drug uptake through porins can be limited by two ways: reduction in the number of porins and mutations [107]. Porin mutations could be achieved by three general processes, shift in the type of pore expressed, change in the level of pore expression, impairment of pore function. Antibiotic resistance, generated by any of these mechanisms responsible for changes in porin permeability, is often associated with other resistance mechanisms, such as increased expression of efflux pumps (described below) [41].
- (b)
- Efflux pumps are active transport proteins involved in the extrusion of substrates against a concentration gradient, including antibiotics, from inside to outside cells [41,108]. Since they rely on energy sources for the active transport of substances from inside to outside the cells, a first general classification divides them based on the mechanism by which they obtain this energy. Primary efflux pumps obtain energy from the active hydrolysis of ATP, while secondary efflux pumps obtain energy from chemical gradients formed by protons or ions such as sodium [109]. First described in the 1990s as a mechanism of drug resistance, numerous efflux pumps in bacteria have subsequently been characterized through molecular biology studies. Five major families of efflux pumps have been described in prokaryotes, namely: (i) adenosine triphosphate binding cassettes (ABCs), primary active transporters that utilize energy derived from ATP hydrolysis; (ii) small family of multidrug resistance (SMR family), unusually small proteins that are predicted to cross the membrane only four times; (iii) multidrug and toxin extrusion (MATE) family that uses a Na+ gradient as an energy source; (iv) major facilitator superfamily (MFS) that uses solute/cation (H+ or Na+) symporter or solute/H+ antiporter; (v) nodular resistance cell division (RND) family, capable of exploiting the substrate/H+ antiporter mechanism and involved in the efflux of multiple antibiotics simultaneously, being multidrug transporters, as well as molecules of detergents, dyes, heavy metals, solvents and many other substrates [70]. These families are classified according to the differences shown in their structural conformation, energy source, range of substrates they can extrude, and type of bacteria in which they are distributed.
- (iii)
- Modifications and/or bypasses of target sites
- (iv)
- Global cellular adaptive process
6. Classification and Evolution of Resistance Genes
7. Development of Resistance Genes in Different Environments and the Part Played by Humans
8. Hypotheses for Solutions to Antibiotic Resistance
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Halawa, E.M.; Fadel, M.; Al-Rabia, M.W.; Behairy, A.; Nouh, N.A.; Abdo, M.; Olga, R.; Fericean, L.; Atwa, A.M.; El-Nablaway, M.; et al. Antibiotic action and resistance: Updated review of mechanisms, spread, influencing factors, and alternative approaches for combating resistance. Front. Pharmacol. 2023, 14, 1305294. [Google Scholar] [CrossRef] [PubMed]
- Matthew, I.H.; Andrew, W.T.; Barrie, W. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Peterson, S.B.; Bertolli, S.K.; Mougous, J.D. The Central Role of Interbacterial Antagonism in Bacterial Life. Curr. Biol. 2020, 30, R1203–R1214. [Google Scholar] [CrossRef] [PubMed]
- Lobanovska, M.; Pilla, G. Penicillin’s Discovery and Antibiotic Resistance: Lessons for the Future? Yale J. Biol. Med. 2017, 90, 135–145. [Google Scholar]
- Selwyn, S. Pioneer work on the ’penicillin phenomenon’, 1870–1876. J. Antimicrob. Chemother. 1979, 5, 249–255. [Google Scholar] [CrossRef]
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef]
- Aminov, R.I.; Mackie, R.I. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef]
- Cantas, L.; Shah, S.Q.; Cavaco, L.M.; Manaia, C.M.; Walsh, F.; Popowska, M.; Garelick, H.; Burgmann, H.; Sorum, H. A brief multi-disciplinary review on antimicrobial resistance in medicine and its linkage to the global environmental microbiota. Front. Microbiol. 2013, 4, 96. [Google Scholar] [CrossRef]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18. [Google Scholar] [CrossRef]
- Huemer, M.; Mairpady Shambat, S.; Brugger, S.D.; Zinkernagel, A.S. Antibiotic resistance and persistence-Implications for human health and treatment perspectives. EMBO Rep. 2020, 21, e51034. [Google Scholar] [CrossRef] [PubMed]
- Gladys, L.H.; Karl, M.; Eleanor, C. Observations on the Mechanism of Action of Penicillin. Proc. Soc. Exp. Biol. Med. 1942, 50, 281–285. [Google Scholar] [CrossRef]
- Bigger, J. Treatment of Staphylococcal Infections with Penicillin by Intermittent Sterilisation. Lancet 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Windels, E.M.; Michiels, J.E.; Fauvart, M.; Wenseleers, T.; Van den Bergh, B.; Michiels, J. Bacterial persistence promotes the evolution of antibiotic resistance by increasing survival and mutation rates. ISME J. 2019, 13, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Niu, H.; Shi, W.; Zhang, S.; Zhang, W.; Zhang, Y. Identification of Genes Involved in Bacteriostatic Antibiotic-Induced Persister Formation. Front. Microbiol. 2018, 9, 413. [Google Scholar] [CrossRef]
- Naghavi, M.; Vollset, S.E.; Ikuta, K.S.; Swetschinski, L.R.; Gray, A.P.; Wool, E.E.; Aguilar, G.R.; Mestrovic, T.; Smith, G.; Han, C. Global burden of bacterial antimicrobial resistance 1990–2021: A systematic analysis with forecasts to 2050. Lancet 2024, 404, 1199–1226. [Google Scholar] [CrossRef]
- World Health Organization. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2022; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- Kosiyaporn, H.; Chanvatik, S.; Issaramalai, T.; Kaewkhankhaeng, W.; Kulthanmanusorn, A.; Saengruang, N.; Witthayapipopsakul, W.; Viriyathorn, S.; Kirivan, S.; Kunpeuk, W.; et al. Surveys of knowledge and awareness of antibiotic use and antimicrobial resistance in general population: A systematic review. PLoS ONE 2020, 15, e0227973. [Google Scholar] [CrossRef]
- O’Neill, J.I.M. Antimicrobial resistance: Tackling a crisis for the health and wealth of nations. In Review on Antimicrobial Resistance; UK Government: London, UK, 2014. [Google Scholar]
- Llor, C.; Bjerrum, L. Antimicrobial resistance: Risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf. 2014, 5, 229–241. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef]
- Dadgostar, P. Antimicrobial Resistance: Implications and Costs. Infect. Drug Resist. 2019, 12, 3903–3910. [Google Scholar] [CrossRef]
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; 9789240026438 (Electronic Version), 9789240026421 (Print Version), 9789240026384 (версия oнлайн), 9789240026391 (версия для печати); World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Antibiotic resistance. Br. Dent. J. 2017, 223, 692. [CrossRef] [PubMed]
- Chokshi, A.; Sifri, Z.; Cennimo, D.; Horng, H. Global Contributors to Antibiotic Resistance. J. Glob. Infect. Dis. 2019, 11, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Rehman, M.T.; Shahwan, M.; Aatif, M. Origin of Antibiotics and Antibiotic Resistance, and Their Impacts on Drug Development: A Narrative Review. Pharmaceuticals 2023, 16, 1615. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D.; Poinar, H. Antibiotic resistance is ancient: Implications for drug discovery. Trends Microbiol. 2012, 20, 157–159. [Google Scholar] [CrossRef]
- Segawa, T.; Takeuchi, N.; Rivera, A.; Yamada, A.; Yoshimura, Y.; Barcaza, G.; Shinbori, K.; Motoyama, H.; Kohshima, S.; Ushida, K. Distribution of antibiotic resistance genes in glacier environments. Environ. Microbiol. Rep. 2013, 5, 127–134. [Google Scholar] [CrossRef]
- Pawlowski, A.C.; Wang, W.; Koteva, K.; Barton, H.A.; McArthur, A.G.; Wright, G.D. A diverse intrinsic antibiotic resistome from a cave bacterium. Nat. Commun. 2016, 7, 13803. [Google Scholar] [CrossRef]
- Shen, Y.; Stedtfeld, R.D.; Guo, X.; Bhalsod, G.D.; Jeon, S.; Tiedje, J.M.; Li, H.; Zhang, W. Pharmaceutical exposure changed antibiotic resistance genes and bacterial communities in soil-surface- and overhead-irrigated greenhouse lettuce. Environ. Int. 2019, 131, 105031. [Google Scholar] [CrossRef]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The evolving role of chemical synthesis in antibacterial drug discovery. Angew. Chem. Int. Ed. Engl. 2014, 53, 8840–8869. [Google Scholar] [CrossRef]
- Bahram, M.; Hildebrand, F.; Forslund, S.K.; Anderson, J.L.; Soudzilovskaia, N.A.; Bodegom, P.M.; Bengtsson-Palme, J.; Anslan, S.; Coelho, L.P.; Harend, H.; et al. Structure and function of the global topsoil microbiome. Nature 2018, 560, 233–237. [Google Scholar] [CrossRef]
- Nguyen, B.T.; Chen, Q.L.; He, J.Z.; Hu, H.W. Microbial regulation of natural antibiotic resistance: Understanding the protist-bacteria interactions for evolution of soil resistome. Sci. Total Environ. 2020, 705, 135882. [Google Scholar] [CrossRef]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.; Wright, G.D. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013, 303, 287–292. [Google Scholar] [CrossRef]
- Jian, Z.; Zeng, L.; Xu, T.; Sun, S.; Yan, S.; Yang, L.; Huang, Y.; Jia, J.; Dou, T. Antibiotic resistance genes in bacteria: Occurrence, spread, and control. J. Basic Microbiol. 2021, 61, 1049–1070. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, R.; Gerhardt, P. Molecular sieving by the Bacillus megaterium cell wall and protoplast. J. Bacteriol. 1971, 107, 718–735. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Antimicrobial resistance in gram-positive bacteria. Am. J. Infect. Control 2006, 34, S11–S19; discussion S64–S73. [Google Scholar] [CrossRef]
- Karaman, R.; Jubeh, B.; Breijyeh, Z. Resistance of Gram-Positive Bacteria to Current Antibacterial Agents and Overcoming Approaches. Molecules 2020, 25, 2888. [Google Scholar] [CrossRef]
- Chancey, S.T.; Zahner, D.; Stephens, D.S. Acquired inducible antimicrobial resistance in Gram-positive bacteria. Future Microbiol. 2012, 7, 959–978. [Google Scholar] [CrossRef]
- Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev. 1992, 56, 395–411. [Google Scholar] [CrossRef]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Barton, H.A.; Wright, G.D. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS ONE 2012, 7, e34953. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Coculescu, B.I. Antimicrobial resistance induced by genetic changes. J. Med. Life 2009, 2, 114–123. [Google Scholar] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef]
- Blazquez, J.; Couce, A.; Rodriguez-Beltran, J.; Rodriguez-Rojas, A. Antimicrobials as promoters of genetic variation. Curr. Opin. Microbiol. 2012, 15, 561–569. [Google Scholar] [CrossRef]
- Stevenson, C.; Hall, J.P.; Harrison, E.; Wood, A.; Brockhurst, M.A. Gene mobility promotes the spread of resistance in bacterial populations. ISME J. 2017, 11, 1930–1932. [Google Scholar] [CrossRef]
- Humphrey, M.; Larrouy-Maumus, G.J.; Furniss, R.C.D.; Mavridou, D.A.I.; Sabnis, A.; Edwards, A.M. Colistin resistance in Escherichia coli confers protection of the cytoplasmic but not outer membrane from the polymyxin antibiotic. Microbiology 2021, 167, 001104. [Google Scholar] [CrossRef]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef]
- Chambers, D.R.; Carleton, W.T.; McEnally, R.W. Immunizing Default-Free Bond Portfolios with a Duration Vector. J. Financ. Quant. Anal. 1988, 23, 89–104. [Google Scholar] [CrossRef]
- Smillie, C.; Garcillan-Barcia, M.P.; Francia, M.V.; Rocha, E.P.; de la Cruz, F. Mobility of plasmids. Microbiol. Mol. Biol. Rev. 2010, 74, 434–452. [Google Scholar] [CrossRef]
- Chaconas, G.; Kobryn, K. Structure, function, and evolution of linear replicons in Borrelia. Annu. Rev. Microbiol. 2010, 64, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C. Update on macrolide-lincosamide-streptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol. Lett. 2008, 282, 147–159. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Pu, W.; Liu, X.; Zhang, Z.; Han, M.; Li, Y.; Huang, X.; Han, X.; Li, Y.; Liu, K.; et al. Proliferation tracing reveals regional hepatocyte generation in liver homeostasis and repair. Science 2021, 371, abc4346. [Google Scholar] [CrossRef] [PubMed]
- Bhat, B.A.; Mir, R.A.; Qadri, H.; Dhiman, R.; Almilaibary, A.; Alkhanani, M.; Mir, M.A. Integrons in the development of antimicrobial resistance: Critical review and perspectives. Front. Microbiol. 2023, 14, 1231938. [Google Scholar] [CrossRef]
- Sandoval-Quintana, E.; Lauga, B.; Cagnon, C. Environmental integrons: The dark side of the integron world. Trends Microbiol. 2023, 31, 432–434. [Google Scholar] [CrossRef]
- Depardieu, F.; Podglajen, I.; Leclercq, R.; Collatz, E.; Courvalin, P. Modes and modulations of antibiotic resistance gene expression. Clin. Microbiol. Rev. 2007, 20, 79–114. [Google Scholar] [CrossRef]
- Mazel, D. Integrons: Agents of bacterial evolution. Nat. Rev. Microbiol. 2006, 4, 608–620. [Google Scholar] [CrossRef]
- Carattoli, A. Importance of integrons in the diffusion of resistance. Vet. Res. 2001, 32, 243–259. [Google Scholar] [CrossRef]
- Partridge, S.R.; Tsafnat, G.; Coiera, E.; Iredell, J.R. Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol. Rev. 2009, 33, 757–784. [Google Scholar] [CrossRef]
- Fluit, A.C.; Schmitz, F.J. Resistance integrons and super-integrons. Clin. Microbiol. Infect. 2004, 10, 272–288. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Thurtle-Schmidt, D.M.; Lo, T.W. Molecular biology at the cutting edge: A review on CRISPR/CAS9 gene editing for undergraduates. Biochem. Mol. Biol. Educ. 2018, 46, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.A.; Wertz, J.E. Bacteriocins: Evolution, ecology, and application. Annu. Rev. Microbiol. 2002, 56, 117–137. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.A.; Roberts, G.A.; Johnson, K.A.; Cooper, L.P.; Liu, H.; White, J.H.; Carter, L.G.; Sanghvi, B.; Oke, M.; Walkinshaw, M.D.; et al. Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 2009, 37, 4887–4897. [Google Scholar] [CrossRef]
- Morar, M.; Wright, G.D. The genomic enzymology of antibiotic resistance. Annu. Rev. Genet. 2010, 44, 25–51. [Google Scholar] [CrossRef]
- Blair, J.M.A. A climate for antibiotic resistance. Nat. Clim. Change 2018, 8, 460–461. [Google Scholar] [CrossRef]
- Price, R. O’Neill report on antimicrobial resistance: Funding for antimicrobial specialists should be improved. Eur. J. Hosp. Pharm. 2016, 23, 245–247. [Google Scholar] [CrossRef]
- Blair, J.M.; Richmond, G.E.; Piddock, L.J. Multidrug efflux pumps in Gram-negative bacteria and their role in antibiotic resistance. Future Microbiol. 2014, 9, 1165–1177. [Google Scholar] [CrossRef]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef]
- Schwarz, S.; Shen, J.; Kadlec, K.; Wang, Y.; Brenner Michael, G.; Fessler, A.T.; Vester, B. Lincosamides, Streptogramins, Phenicols, and Pleuromutilins: Mode of Action and Mechanisms of Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a027037. [Google Scholar] [CrossRef]
- Egorov, A.M.; Ulyashova, M.M.; Rubtsova, M.Y. Bacterial Enzymes and Antibiotic Resistance. Acta Nat. 2018, 10, 33–48. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updat. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, N.; Wang, M.; Luo, M.; Peng, Y.; Li, Z.; Xu, J.; Ou, M.; Kan, B.; Li, X. The prevalence and distribution of aminoglycoside resistance genes. Biosaf. Health 2023, 5, 14–20. [Google Scholar] [CrossRef]
- Moussa, A.A.; Md Nordin, A.F.; Hamat, R.A.; Jasni, A.S. High Level Aminoglycoside Resistance and Distribution of the Resistance Genes in Enterococcus faecalis and Enterococcus faecium from Teaching Hospital in Malaysia. Infect Drug Resist. 2019, 12, 3269–3274. [Google Scholar] [CrossRef]
- Galopin, S.; Cattoir, V.; Leclercq, R. A chromosomal chloramphenicol acetyltransferase determinant from a probiotic strain of Bacillus clausii. FEMS Microbiol. Lett. 2009, 296, 185–189. [Google Scholar] [CrossRef]
- Pettis, G.S. Spreading the news about the novel conjugation mechanism in Streptomyces bacteria. Environ. Microbiol. Rep. 2018, 10, 503–510. [Google Scholar] [CrossRef]
- Lund, D.; Kieffer, N.; Parras-Molto, M.; Ebmeyer, S.; Berglund, F.; Johnning, A.; Larsson, D.G.J.; Kristiansson, E. Large-scale characterization of the macrolide resistome reveals high diversity and several new pathogen-associated genes. Microb. Genom. 2022, 8, 000770. [Google Scholar] [CrossRef]
- Fyfe, C.; Grossman, T.H.; Kerstein, K.; Sutcliffe, J. Resistance to Macrolide Antibiotics in Public Health Pathogens. Cold Spring Harb. Perspect. Med. 2016, 6, a025395. [Google Scholar] [CrossRef]
- Lee, C.G.; Lim, Y.J.; Park, S.J.; Jang, B.I.; Choi, S.R.; Kim, J.K.; Kim, Y.T.; Cho, J.Y.; Yang, C.H.; Chun, H.J.; et al. The clinical features and treatment modality of esophageal neuroendocrine tumors: A multicenter study in Korea. BMC Cancer 2014, 14, 569. [Google Scholar] [CrossRef]
- Beharry, Z.; Palzkill, T. Functional analysis of active site residues of the fosfomycin resistance enzyme FosA from Pseudomonas aeruginosa. J. Biol. Chem. 2005, 280, 17786–17791. [Google Scholar] [CrossRef]
- Roberts, A.A.; Sharma, S.V.; Strankman, A.W.; Duran, S.R.; Rawat, M.; Hamilton, C.J. Mechanistic studies of FosB: A divalent-metal-dependent bacillithiol-S-transferase that mediates fosfomycin resistance in Staphylococcus aureus. Biochem. J. 2013, 451, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Fillgrove, K.L.; Pakhomova, S.; Newcomer, M.E.; Armstrong, R.N. Mechanistic diversity of fosfomycin resistance in pathogenic microorganisms. J. Am. Chem. Soc. 2003, 125, 15730–15731. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Kuzuyama, T.; Seto, H. Characterization of the fomA and fomB gene products from Streptomyces wedmorensis, which confer fosfomycin resistance on Escherichia coli. Antimicrob. Agents Chemother. 2000, 44, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Baysarowich, J.; Koteva, K.; Hughes, D.W.; Ejim, L.; Griffiths, E.; Zhang, K.; Junop, M.; Wright, G.D. Rifamycin antibiotic resistance by ADP-ribosylation: Structure and diversity of Arr. Proc. Natl. Acad. Sci. USA 2008, 105, 4886–4891. [Google Scholar] [CrossRef]
- Spanogiannopoulos, P.; Thaker, M.; Koteva, K.; Waglechner, N.; Wright, G.D. Characterization of a rifampin-inactivating glycosyltransferase from a screen of environmental actinomycetes. Antimicrob. Agents Chemother. 2012, 56, 5061–5069. [Google Scholar] [CrossRef]
- Spanogiannopoulos, P.; Waglechner, N.; Koteva, K.; Wright, G.D. A rifamycin inactivating phosphotransferase family shared by environmental and pathogenic bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 7102–7107. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Koteva, K.; Cox, G.; Kelso, J.K.; Surette, M.D.; Zubyk, H.L.; Ejim, L.; Stogios, P.; Savchenko, A.; Sorensen, D.; Wright, G.D. Rox, a Rifamycin Resistance Enzyme with an Unprecedented Mechanism of Action. Cell Chem. Biol. 2018, 25, 403–412 e405. [Google Scholar] [CrossRef]
- Zhu, B.; Yin, H. Alginate lyase: Review of major sources and classification, properties, structure-function analysis and applications. Bioengineered 2015, 6, 125–131. [Google Scholar] [CrossRef]
- Bonomo, R.A. beta-Lactamases: A Focus on Current Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a025239. [Google Scholar] [CrossRef]
- Oelschlaeger, P. beta-Lactamases: Sequence, Structure, Function, and Inhibition. Biomolecules 2021, 11, 986. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. The ABCD’s of beta-lactamase nomenclature. J. Infect. Chemother. 2013, 19, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Fatima, J.; Shakil, S.; Rizvi, S.M.; Kamal, M.A. Antibiotic resistance and extended spectrum beta-lactamases: Types, epidemiology and treatment. Saudi J. Biol. Sci. 2015, 22, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef]
- Bradford, P.A.; Bratu, S.; Urban, C.; Visalli, M.; Mariano, N.; Landman, D.; Rahal, J.J.; Brooks, S.; Cebular, S.; Quale, J. Emergence of carbapenem-resistant Klebsiella species possessing the class A carbapenem-hydrolyzing KPC-2 and inhibitor-resistant TEM-30 beta-lactamases in New York City. Clin. Infect. Dis. 2004, 39, 55–60. [Google Scholar] [CrossRef]
- Orencia, M.C.; Yoon, J.S.; Ness, J.E.; Stemmer, W.P.; Stevens, R.C. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Struct. Biol. 2001, 8, 238–242. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC beta-lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef]
- Yoon, E.J.; Jeong, S.H. Class D β-lactamases. J. Antimicrob. Chemother. 2021, 76, 836–864. [Google Scholar] [CrossRef]
- Bebrone, C. Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 2007, 74, 1686–1701. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L.; Walsh, T.R.; Livermore, D.M. The emerging NDM carbapenemases. Trends Microbiol. 2011, 19, 588–595. [Google Scholar] [CrossRef]
- Gomes, C.; Martínez-Puchol, S.; Palma, N.; Horna, G.; Ruiz-Roldán, L.; Pons, M.J.; Ruiz, J. Macrolide resistance mechanisms in Enterobacteriaceae: Focus on azithromycin. Crit. Rev. Microbiol. 2017, 43, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Barkhouse, A.; Hackenberger, D.; Wright, G.D. Antibiotic resistance: A key microbial survival mechanism that threatens public health. Cell Host Microbe 2024, 32, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.J.; Simjee, S.; Al-Hattawi, K.; Robertson, B.D.; Easmon, C.S.; Ison, C.A. Gonococcal resistance to beta-lactams and tetracycline involves mutation in loop 3 of the porin encoded at the penB locus. Antimicrob. Agents Chemother. 1998, 42, 2799–2803. [Google Scholar] [CrossRef] [PubMed]
- Costa-Riu, N.; Maier, E.; Burkovski, A.; Krämer, R.; Lottspeich, F.; Benz, R. Identification of an anion-specific channel in the cell wall of the Gram-positive bacterium Corynebacterium glutamicum. Mol. Microbiol. 2003, 50, 1295–1308. [Google Scholar] [CrossRef]
- Kumar, A.; Schweizer, H.P. Bacterial resistance to antibiotics: Active efflux and reduced uptake. Adv. Drug Deliv. Rev. 2005, 57, 1486–1513. [Google Scholar] [CrossRef]
- Piddock, L.J. Multidrug-resistance efflux pumps—Not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, V.K.; Pathania, R. Efflux pump inhibitors for bacterial pathogens: From bench to bedside. Indian J. Med. Res. 2019, 149, 129–145. [Google Scholar] [CrossRef]
- Wilson, D.N.; Hauryliuk, V.; Atkinson, G.C.; O’Neill, A.J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 2020, 18, 637–648. [Google Scholar] [CrossRef]
- Manavathu, E.K.; Fernandez, C.L.; Cooperman, B.S.; Taylor, D.E. Molecular studies on the mechanism of tetracycline resistance mediated by Tet(O). Antimicrob. Agents Chemother. 1990, 34, 71–77. [Google Scholar] [CrossRef]
- Burdett, V. Tet(M)-promoted release of tetracycline from ribosomes is GTP dependent. J. Bacteriol. 1996, 178, 3246–3251. [Google Scholar] [CrossRef]
- Connell, S.R.; Tracz, D.M.; Nierhaus, K.H.; Taylor, D.E. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob. Agents Chemother. 2003, 47, 3675–3681. [Google Scholar] [CrossRef] [PubMed]
- Donhofer, A.; Franckenberg, S.; Wickles, S.; Berninghausen, O.; Beckmann, R.; Wilson, D.N. Structural basis for TetM-mediated tetracycline resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 16900–16905. [Google Scholar] [CrossRef]
- Li, W.; Atkinson, G.C.; Thakor, N.S.; Allas, U.; Lu, C.C.; Chan, K.Y.; Tenson, T.; Schulten, K.; Wilson, K.S.; Hauryliuk, V.; et al. Mechanism of tetracycline resistance by ribosomal protection protein Tet(O). Nat. Commun. 2013, 4, 1477. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martinez, J.M.; Cano, M.E.; Velasco, C.; Martinez-Martinez, L.; Pascual, A. Plasmid-mediated quinolone resistance: An update. J. Infect. Chemother. 2011, 17, 149–182. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martinez, L.; Eliecer Cano, M.; Manuel Rodriguez-Martinez, J.; Calvo, J.; Pascual, A. Plasmid-mediated quinolone resistance. Expert Rev. Anti Infect. Ther. 2008, 6, 685–711. [Google Scholar] [CrossRef]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef]
- Lambert, P.A. Bacterial resistance to antibiotics: Modified target sites. Adv. Drug Deliv. Rev. 2005, 57, 1471–1485. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W. Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- Hooper, D.C. Fluoroquinolone resistance among Gram-positive cocci. Lancet Infect. Dis. 2002, 2, 530–538. [Google Scholar] [CrossRef]
- Mendes, R.E.; Deshpande, L.M.; Jones, R.N. Linezolid update: Stable in vitro activity following more than a decade of clinical use and summary of associated resistance mechanisms. Drug Resist. Updat. 2014, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Schaenzer, A.J.; Wright, G.D. Antibiotic Resistance by Enzymatic Modification of Antibiotic Targets. Trends Mol. Med. 2020, 26, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, R. Mechanisms of resistance to macrolides and lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4, 1221–1236. [Google Scholar] [CrossRef]
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef]
- Flensburg, J.; Skold, O. Massive overproduction of dihydrofolate reductase in bacteria as a response to the use of trimethoprim. Eur. J. Biochem. 1987, 162, 473–476. [Google Scholar] [CrossRef]
- Huovinen, P. Resistance to trimethoprim-sulfamethoxazole. Clin. Infect. Dis. 2001, 32, 1608–1614. [Google Scholar] [CrossRef]
- Zervos, M.J.; Schaberg, D.R. Reversal of the in vitro susceptibility of enterococci to trimethoprim-sulfamethoxazole by folinic acid. Antimicrob. Agents Chemother. 1985, 28, 446–448. [Google Scholar] [CrossRef]
- Bourne, C.R. Utility of the Biosynthetic Folate Pathway for Targets in Antimicrobial Discovery. Antibiotics 2014, 3, 1. [Google Scholar] [CrossRef]
- Rodrigo, M.K.D.; Saiganesh, A.; Hayes, A.J.; Wilson, A.M.; Anstey, J.; Pickering, J.L.; Iwasaki, J.; Hillas, J.; Winslow, S.; Woodman, T.; et al. Host-dependent resistance of Group A Streptococcus to sulfamethoxazole mediated by a horizontally-acquired reduced folate transporter. Nat. Commun. 2022, 13, 6557. [Google Scholar] [CrossRef]
- Raza, S.; Matuła, K.; Karoń, S.; Paczesny, J. Resistance and Adaptation of Bacteria to Non-Antibiotic Antibacterial Agents: Physical Stressors, Nanoparticles, and Bacteriophages. Antibiotics 2021, 10, 435. [Google Scholar] [CrossRef] [PubMed]
- Kawasuji, H.; Nagaoka, K.; Tsuji, Y.; Kimoto, K.; Takegoshi, Y.; Kaneda, M.; Murai, Y.; Karaushi, H.; Mitsutake, K.; Yamamoto, Y. Effectiveness and Safety of Linezolid Versus Vancomycin, Teicoplanin, or Daptomycin against Methicillin-Resistant Staphylococcus aureus Bacteremia: A Systematic Review and Meta-Analysis. Antibiotics 2023, 12, 697. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Muraih, J.K.; Tishbi, N.; Herskowitz, J.; Victor, R.L.; Silverman, J.; Uwumarenogie, S.; Taylor, S.D.; Palmer, M.; Mintzer, E. Cardiolipin prevents membrane translocation and permeabilization by daptomycin. J. Biol. Chem. 2014, 289, 11584–11591. [Google Scholar] [CrossRef] [PubMed]
- Roy, H.; Dare, K.; Ibba, M. Adaptation of the bacterial membrane to changing environments using aminoacylated phospholipids. Mol. Microbiol. 2009, 71, 547–550. [Google Scholar] [CrossRef]
- Howden, B.P.; Davies, J.K.; Johnson, P.D.; Stinear, T.P.; Grayson, M.L. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: Resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 2010, 23, 99–139. [Google Scholar] [CrossRef]
- Gardete, S.; Tomasz, A. Mechanisms of vancomycin resistance in Staphylococcus aureus. J. Clin. Investig. 2014, 124, 2836–2840. [Google Scholar] [CrossRef]
- Schulze, A.; Mitterer, F.; Pombo, J.P.; Schild, S. Biofilms by bacterial human pathogens: Clinical relevance—Development, composition and regulation—Therapeutical strategies. Microb. Cell 2021, 8, 28–56. [Google Scholar] [CrossRef]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial Biofilm: A Review on Formation, Infection, Antibiotic Resistance, Control Measures, and Innovative Treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef]
- Lopatkin, A.J.; Huang, S.; Smith, R.P.; Srimani, J.K.; Sysoeva, T.A.; Bewick, S.; Karig, D.K.; You, L. Antibiotics as a selective driver for conjugation dynamics. Nat. Microbiol. 2016, 1, 16044. [Google Scholar] [CrossRef]
- Molnar, A. Antimicrobial Resistance Awareness and Games. Trends Microbiol. 2019, 27, 1–3. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T. Infective heredity of multiple drug resistance in bacteria. Bacteriol. Rev. 1963, 27, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C. Antibiotic toxicity, interactions and resistance development. Periodontol 2000 2002, 28, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Skold, O. Resistance to trimethoprim and sulfonamides. Vet. Res. 2001, 32, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Zafar, W.; Sumrra, S.H.; Hassan, A.U.; Chohan, Z.H. A review on ‘sulfonamides’: Their chemistry and pharmacological potentials for designing therapeutic drugs in medical science. J. Coord. Chem. 2023, 76, 546–580. [Google Scholar] [CrossRef]
- Venkatesan, M.; Fruci, M.; Verellen, L.A.; Skarina, T.; Mesa, N.; Flick, R.; Pham, C.; Mahadevan, R.; Stogios, P.J.; Savchenko, A. Molecular mechanism of plasmid-borne resistance to sulfonamide antibiotics. Nat. Commun. 2023, 14, 4031. [Google Scholar] [CrossRef]
- Radstrom, P.; Swedberg, G. RSF1010 and a conjugative plasmid contain sulII, one of two known genes for plasmid-borne sulfonamide resistance dihydropteroate synthase. Antimicrob. Agents Chemother. 1988, 32, 1684–1692. [Google Scholar] [CrossRef]
- Perreten, V.; Boerlin, P. A new sulfonamide resistance gene (sul3) in Escherichia coli is widespread in the pig population of Switzerland. Antimicrob. Agents Chemother. 2003, 47, 1169–1172. [Google Scholar] [CrossRef]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile beta-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef]
- Sanchez, M.B. Antibiotic resistance in the opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 2015, 6, 658. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, R.A.; Szabo, D. Mechanisms of multidrug resistance in Acinetobacter species and Pseudomonas aeruginosa. Clin. Infect. Dis. 2006, 43 (Suppl. S2), S49–S56. [Google Scholar] [CrossRef] [PubMed]
- Peric, M.; Bozdogan, B.; Jacobs, M.R.; Appelbaum, P.C. Effects of an efflux mechanism and ribosomal mutations on macrolide susceptibility of Haemophilus influenzae clinical isolates. Antimicrob. Agents Chemother. 2003, 47, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Kakoullis, L.; Papachristodoulou, E.; Chra, P.; Panos, G. Mechanisms of antibiotic resistance in important gram-positive and gram-negative pathogens and novel antibiotic solutions. Antibiotics 2021, 10, 415. [Google Scholar] [CrossRef]
- Schweizer, H.P. Efflux as a mechanism of resistance to antimicrobials in Pseudomonas aeruginosa and related bacteria: Unanswered questions. Genet. Mol. Res. 2003, 2, 48–62. [Google Scholar]
- Ebbensgaard, A.E.; Lobner-Olesen, A.; Frimodt-Moller, J. The Role of Efflux Pumps in the Transition from Low-Level to Clinical Antibiotic Resistance. Antibiotics 2020, 9, 855. [Google Scholar] [CrossRef]
- Poirel, L.; Naas, T.; Nordmann, P. Diversity, epidemiology, and genetics of class D beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 24–38. [Google Scholar] [CrossRef]
- Nordmann, P.; Gniadkowski, M.; Giske, C.G.; Poirel, L.; Woodford, N.; Miriagou, V.; European Network on Carbapenemases. Identification and screening of carbapenemase-producing Enterobacteriaceae. Clin. Microbiol. Infect. 2012, 18, 432–438. [Google Scholar] [CrossRef]
- Walsh, M. Multimodal literacy: What does it mean for classroom practice? Aust. J. Lang. Lit. 2010, 33, 211–239. [Google Scholar] [CrossRef]
- McCormick, M.H.; McGuire, J.M.; Pittenger, G.E.; Pittenger, R.C.; Stark, W.M. Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot. Annu. 1955, 3, 606–611. [Google Scholar]
- Weisblum, B. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 1995, 39, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Kotra, L.P.; Haddad, J.; Mobashery, S. Aminoglycosides: Perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 2000, 44, 3249–3256. [Google Scholar] [CrossRef] [PubMed]
- Begg, E.J.; Barclay, M.L.; Duffull, S.B. A suggested approach to once-daily aminoglycoside dosing. Br. J. Clin. Pharmacol. 1995, 39, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Hooper, D.C. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin. Infect. Dis. 2000, 31 (Suppl. S2), S24–S28. [Google Scholar] [CrossRef]
- King, D.E.; Malone, R.; Lilley, S.H. New classification and update on the quinolone antibiotics. Am. Fam. Physician 2000, 61, 2741–2748. [Google Scholar]
- Courvalin, P. New plasmid-mediated resistances to antimicrobial agents. Arch. Microbiol. 2008, 189, 289–291. [Google Scholar] [CrossRef]
- Jacoby, G.A. Mechanisms of resistance to quinolones. Clin. Infect. Dis. 2005, 41 (Suppl. S2), S120–S126. [Google Scholar] [CrossRef]
- Strahilevitz, J.; Jacoby, G.A.; Hooper, D.C.; Robicsek, A. Plasmid-mediated quinolone resistance: A multifaceted threat. Clin. Microbiol. Rev. 2009, 22, 664–689. [Google Scholar] [CrossRef]
- Cattoir, V.; Poirel, L.; Nordmann, P. Plasmid-mediated quinolone resistance pump QepA2 in an Escherichia coli isolate from France. Antimicrob. Agents Chemother. 2008, 52, 3801–3804. [Google Scholar] [CrossRef]
- Perichon, B.; Courvalin, P.; Galimand, M. Transferable resistance to aminoglycosides by methylation of G1405 in 16S rRNA and to hydrophilic fluoroquinolones by QepA-mediated efflux in Escherichia coli. Antimicrob. Agents Chemother. 2007, 51, 2464–2469. [Google Scholar] [CrossRef]
- Li, J.; Nation, R.L.; Turnidge, J.D.; Milne, R.W.; Coulthard, K.; Rayner, C.R.; Paterson, D.L. Colistin: The re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 2006, 6, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Poudyal, A.; Howden, B.P.; Bell, J.M.; Gao, W.; Owen, R.J.; Turnidge, J.D.; Nation, R.L.; Li, J. In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J. Antimicrob. Chemother. 2008, 62, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Li, J.; Rayner, C.R.; Nation, R.L. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Beno, P.; Krcmery, V.; Demitrovicova, A. Bacteraemia in cancer patients caused by colistin-resistant Gram-negative bacilli after previous exposure to ciprofloxacin and/or colistin. Clin. Microbiol. Infect. 2006, 12, 497–498. [Google Scholar] [CrossRef]
- Antoniadou, A.; Kontopidou, F.; Poulakou, G.; Koratzanis, E.; Galani, I.; Papadomichelakis, E.; Kopterides, P.; Souli, M.; Armaganidis, A.; Giamarellou, H. Colistin-resistant isolates of Klebsiella pneumoniae emerging in intensive care unit patients: First report of a multiclonal cluster. J. Antimicrob. Chemother. 2007, 59, 786–790. [Google Scholar] [CrossRef]
- Sultan, I.; Rahman, S.; Jan, A.T.; Siddiqui, M.T.; Mondal, A.H.; Haq, Q.M.R. Antibiotics, Resistome and Resistance Mechanisms: A Bacterial Perspective. Front. Microbiol. 2018, 9, 2066. [Google Scholar] [CrossRef]
- Mohapatra, S.R.; Sadik, A.; Sharma, S.; Poschet, G.; Gegner, H.M.; Lanz, T.V.; Lucarelli, P.; Klingmüller, U.; Platten, M.; Heiland, I.; et al. Hypoxia Routes Tryptophan Homeostasis Towards Increased Tryptamine Production. Front. Immunol. 2021, 12, 590532. [Google Scholar] [CrossRef]
- Liu, J.H.; Liu, Y.Y.; Shen, Y.B.; Yang, J.; Walsh, T.R.; Wang, Y.; Shen, J. Plasmid-mediated colistin-resistance genes: Mcr. Trends Microbiol. 2024, 32, 365–378. [Google Scholar] [CrossRef]
- Rubens, R.S.; Arruda, I.S.A.; Almeida, R.M.; Nobrega, Y.K.M.; Carneiro, M.D.S.; Dalmolin, T.V. Challenges in the Detection of Polymyxin Resistance: From Today to the Future. Microorganisms 2024, 12, 101. [Google Scholar] [CrossRef]
- Yao, R.; Crandall, L. Glycopeptides: Classification, Occurrence, and Discovery; CRC Press: Boca Raton, FL, USA, 2020; pp. 1–28. [Google Scholar]
- Gao, Y. Glycopeptide antibiotics and development of inhibitors to overcome vancomycin resistance. Nat. Prod. Rep. 2002, 19, 100–107. [Google Scholar] [CrossRef]
- Klare, I.; Konstabel, C.; Badstubner, D.; Werner, G.; Witte, W. Occurrence and spread of antibiotic resistances in Enterococcus faecium. Int. J. Food Microbiol. 2003, 88, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Aarestrup, F.M. Occurrence of glycopeptide resistance among Enterococcus faecium isolates from conventional and ecological poultry farms. Microb. Drug Resist. 1995, 1, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Klare, I.; Heier, H.; Claus, H.; Reissbrodt, R.; Witte, W. vanA-mediated high-level glycopeptide resistance in Enterococcus faecium from animal husbandry. FEMS Microbiol. Lett. 1995, 125, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Selim, S. Mechanisms of gram-positive vancomycin resistance (Review). Biomed. Rep. 2022, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Shan, Y.; Pan, Q.; Gao, X.; Yan, A. Anaerobic expression of the gadE-mdtEF multidrug efflux operon is primarily regulated by the two-component system ArcBA through antagonizing the H-NS mediated repression. Front. Microbiol. 2013, 4, 194. [Google Scholar] [CrossRef]
- Warner, D.M.; Shafer, W.M.; Jerse, A.E. Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE Efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol. Microbiol. 2008, 70, 462–478. [Google Scholar] [CrossRef]
- Webber, M.A.; Talukder, A.; Piddock, L.J. Contribution of mutation at amino acid 45 of AcrR to acrB expression and ciprofloxacin resistance in clinical and veterinary Escherichia coli isolates. Antimicrob. Agents Chemother. 2005, 49, 4390–4392. [Google Scholar] [CrossRef]
- Sahal, G.; Bilkay, I.S. Multi drug resistance in strong biofilm forming clinical isolates of Staphylococcus epidermidis. Braz. J. Microbiol. 2014, 45, 539–544. [Google Scholar] [CrossRef]
- Lawther, K.; Santos, F.G.; Oyama, L.B.; Rubino, F.; Morrison, S.; Creevey, C.J.; McGrath, J.W.; Huws, S.A. Resistome Analysis of Global Livestock and Soil Microbiomes. Front. Microbiol. 2022, 13, 897905. [Google Scholar] [CrossRef]
- Wollein Waldetoft, K.; Sundius, S.; Kuske, R.; Brown, S.P. Defining the Benefits of Antibiotic Resistance in Commensals and the Scope for Resistance Optimization. mBio 2023, 14, e0134922. [Google Scholar] [CrossRef]
- Zhuang, M.; Achmon, Y.; Cao, Y.; Liang, X.; Chen, L.; Wang, H.; Siame, B.A.; Leung, K.Y. Distribution of antibiotic resistance genes in the environment. Environ. Pollut. 2021, 285, 117402. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, T.P.; Glennon, E.E.; Chen, D.; Gilbert, M.; Robinson, T.P.; Grenfell, B.T.; Levin, S.A.; Bonhoeffer, S.; Laxminarayan, R. Reducing antimicrobial use in food animals. Science 2017, 357, 1350–1352. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; White, D.G.; Friedman, S.L.; Glenn, A.; Blickenstaff, K.; Ayers, S.L.; Abbott, J.W.; Hall-Robinson, E.; McDermott, P.F. Antimicrobial resistance in Salmonella enterica serovar Heidelberg isolates from retail meats, including poultry, from 2002 to 2006. Appl. Environ. Microbiol. 2008, 74, 6656–6662. [Google Scholar] [CrossRef]
- Zinsstag, J.; Schelling, E.; Waltner-Toews, D.; Tanner, M. From “one medicine” to “one health” and systemic approaches to health and well-being. Prev. Vet. Med. 2011, 101, 148–156. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, X.; Li, J.; Lv, N.; Liu, F.; Wu, J.; Lin, I.Y.; Wu, N.; Weimer, B.C.; Gao, G.F.; et al. The Bacterial Mobile Resistome Transfer Network Connecting the Animal and Human Microbiomes. Appl. Environ. Microbiol. 2016, 82, 6672–6681. [Google Scholar] [CrossRef]
- Ben, Y.; Fu, C.; Hu, M.; Liu, L.; Wong, M.H.; Zheng, C. Human health risk assessment of antibiotic resistance associated with antibiotic residues in the environment: A review. Environ. Res. 2019, 169, 483–493. [Google Scholar] [CrossRef]
- Marti, R.; Scott, A.; Tien, Y.C.; Murray, R.; Sabourin, L.; Zhang, Y.; Topp, E. Impact of manure fertilization on the abundance of antibiotic-resistant bacteria and frequency of detection of antibiotic resistance genes in soil and on vegetables at harvest. Appl. Environ. Microbiol. 2013, 79, 5701–5709. [Google Scholar] [CrossRef]
- Reynaga, E.; Navarro, M.; Vilamala, A.; Roure, P.; Quintana, M.; Garcia-Nuñez, M.; Figueras, R.; Torres, C.; Lucchetti, G.; Sabrià, M. Prevalence of colonization by methicillin-resistant Staphylococcus aureus ST398 in pigs and pig farm workers in an area of Catalonia, Spain. BMC Infect. Dis. 2016, 16, 716. [Google Scholar] [CrossRef]
- Wang, B.; Yang, X.L.; Li, W.; Zhu, Y.; Ge, X.Y.; Zhang, L.B.; Zhang, Y.Z.; Bock, C.T.; Shi, Z.L. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in China. Virol. J. 2017, 14, 40. [Google Scholar] [CrossRef]
- Huang, J.; Lv, C.; Li, M.; Rahman, T.; Chang, Y.F.; Guo, X.; Song, Z.; Zhao, Y.; Li, Q.; Ni, P.; et al. Carbapenem-resistant Escherichia coli exhibit diverse spatiotemporal epidemiological characteristics across the globe. Commun. Biol. 2024, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Huang, D.; Du, L.; Song, B.; Yin, L.; Chen, Y.; Gao, L.; Li, R.; Huang, H.; Zeng, G. Antibiotic resistance in soil-plant systems: A review of the source, dissemination, influence factors, and potential exposure risks. Sci. Total Environ. 2023, 869, 161855. [Google Scholar] [CrossRef] [PubMed]
- Scaccia, N.; Vaz-Moreira, I.; Manaia, C.M. The risk of transmitting antibiotic resistance through endophytic bacteria. Trends Plant Sci. 2021, 26, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Winckler, C.; Grafe, A. Use of veterinary drugs in intensive animal production. J. Soils Sediments 2001, 1, 66–70. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Zhao, Y.; Zhu, D.; Gillings, M.; Penuelas, J.; Ok, Y.S.; Capon, A.; Banwart, S. Soil biota, antimicrobial resistance and planetary health. Environ. Int. 2019, 131, 105059. [Google Scholar] [CrossRef]
- Ghosh, S.; LaPara, T.M. The effects of subtherapeutic antibiotic use in farm animals on the proliferation and persistence of antibiotic resistance among soil bacteria. ISME J. 2007, 1, 191–203. [Google Scholar] [CrossRef]
- Iwu, C.D.; Korsten, L.; Okoh, A.I. The incidence of antibiotic resistance within and beyond the agricultural ecosystem: A concern for public health. Microbiologyopen 2020, 9, e1035. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Johnson, T.A.; Su, J.Q.; Qiao, M.; Guo, G.X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef]
- Jaimy, S.; Anupama, K.V.; Nidheesh, P.V. Tetracyclines in the environment: An overview on the occurrence, fate, toxicity, detection, removal methods, and sludge management. Sci. Total Environ. 2021, 771, 145291. [Google Scholar] [CrossRef]
- Fouz, N.; Pangesti, K.N.A.; Yasir, M.; Al-Malki, A.L.; Azhar, E.I.; Hill-Cawthorne, G.A.; Abd El Ghany, M. The Contribution of Wastewater to the Transmission of Antimicrobial Resistance in the Environment: Implications of Mass Gathering Settings. Trop. Med. Infect. Dis. 2020, 5, 33. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, R.; Li, J.; Wu, Z.; Yin, W.; Schwarz, S.; Tyrrell, J.M.; Zheng, Y.; Wang, S.; Shen, Z.; et al. Comprehensive resistome analysis reveals the prevalence of NDM and MCR-1 in Chinese poultry production. Nat. Microbiol. 2017, 2, 16260. [Google Scholar] [CrossRef] [PubMed]
- Osman, K.M.; Kappell, A.D.; Elhadidy, M.; ElMougy, F.; El-Ghany, W.A.A.; Orabi, A.; Mubarak, A.S.; Dawoud, T.M.; Hemeg, H.A.; Moussa, I.M.I.; et al. Poultry hatcheries as potential reservoirs for antimicrobial-resistant Escherichia coli: A risk to public health and food safety. Sci. Rep. 2018, 8, 5859. [Google Scholar] [CrossRef] [PubMed]
- Bayko, B.; Michail, S. Microbial air pollution caused by intensive broiler chicken breeding. FEMS Microbiol. Ecol. 1999, 29, 389–392. [Google Scholar] [CrossRef]
- Kena, Q.; Liangliang, W.; Jianju, L.; Bo, L.; Fengyi, Z.; Hang, Y.; Qingliang, Z.; Kun, W. A review of ARGs in WWTPs: Sources, stressors and elimination. Chin. Chem. Lett. 2020, 31, 2603–2613. [Google Scholar]
- Quiros, P.; Colomer-Lluch, M.; Martinez-Castillo, A.; Miro, E.; Argente, M.; Jofre, J.; Navarro, F.; Muniesa, M. Antibiotic resistance genes in the bacteriophage DNA fraction of human fecal samples. Antimicrob. Agents Chemother. 2014, 58, 606–609. [Google Scholar] [CrossRef]
- Moon, K.; Jeon, J.H.; Kang, I.; Park, K.S.; Lee, K.; Cha, C.J.; Lee, S.H.; Cho, J.C. Freshwater viral metagenome reveals novel and functional phage-borne antibiotic resistance genes. Microbiome 2020, 8, 75. [Google Scholar] [CrossRef]
- Li, F.; Liu, J.; Maldonado-Gómez, M.X.; Frese, S.A.; Gänzle, M.G.; Walter, J. Highly accurate and sensitive absolute quantification of bacterial strains in human fecal samples. Microbiome 2024, 12, 168. [Google Scholar] [CrossRef]
- Blanco-Picazo, P.; Roscales, G.; Toribio-Avedillo, D.; Gomez-Gomez, C.; Avila, C.; Balleste, E.; Muniesa, M.; Rodriguez-Rubio, L. Antibiotic Resistance Genes in Phage Particles from Antarctic and Mediterranean Seawater Ecosystems. Microorganisms 2020, 8, 1293. [Google Scholar] [CrossRef]
- Yang, W.C.; Yen, H.J.; Liang, M.L.; Chen, H.H.; Lee, Y.Y.; Chang, F.C.; Lin, S.C.; Wong, T.T.; Hu, Y.W.; Chen, Y.W. Effect of early radiotherapy initiation and high-dose chemotherapy on the prognosis of pediatric atypical teratoid rhabdoid tumors in different age groups. J. Neurooncol. 2020, 147, 619–631. [Google Scholar] [CrossRef]
- Heß, S.; Kneis, D.; Österlund, T.; Li, B.; Kristiansson, E.; Berendonk, T.U. Sewage from Airplanes Exhibits High Abundance and Diversity of Antibiotic Resistance Genes. Environ. Sci. Technol. 2019, 53, 13898–13905. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G. The structure and diversity of human, animal and environmental resistomes. Microbiome 2016, 4, 54. [Google Scholar] [CrossRef] [PubMed]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, J.; Oguri, S.; Nakamura, S.; Hanawa, T.; Fukumoto, T.; Hayashi, Y.; Kawaguchi, K.; Mizutani, Y.; Yao, T.; Akizawa, K.; et al. Ciliates rapidly enhance the frequency of conjugation between Escherichia coli strains through bacterial accumulation in vesicles. Res. Microbiol. 2010, 161, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.T.; Macdonald, R.E.; Tapscott, B.; Nagy, E.; Turner, P.V. Detection of Astrovirus, Rotavirus C, and Hepatitis E Viral RNA in Adult and Juvenile Farmed Mink (Neovison vison). Front. Vet. Sci. 2018, 5, ARTN 132. [Google Scholar] [CrossRef]
- Li, J.; Cao, J.; Zhu, Y.G.; Chen, Q.L.; Shen, F.; Wu, Y.; Xu, S.; Fan, H.; Da, G.; Huang, R.J.; et al. Global Survey of Antibiotic Resistance Genes in Air. Environ. Sci. Technol. 2018, 52, 10975–10984. [Google Scholar] [CrossRef]
- O’Connor, R.; O’Doherty, J.; O’Regan, A.; Dunne, C. Antibiotic use for acute respiratory tract infections (ARTI) in primary care; what factors affect prescribing and why is it important? A narrative review. Ir. J. Med. Sci. 2018, 187, 969–986. [Google Scholar] [CrossRef]
- Asokan, G.V.; Ramadhan, T.; Ahmed, E.; Sanad, H. WHO Global Priority Pathogens List: A Bibliometric Analysis of Medline-PubMed for Knowledge Mobilization to Infection Prevention and Control Practices in Bahrain. Oman Med. J. 2019, 34, 184–193. [Google Scholar] [CrossRef]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- WHO. WHO Global Strategy for Containment of Antimicrobial Resistance; WHO: Geneva, Switzerland, 2001. [Google Scholar]
- European Centre for Disease Prevention and Control and World Health Organization, Regional Office for Europe. Antimicrobial Resistance Surveillance in Europe 2023–2021 Data; European Centre for Disease Prevention and Control and World Health Organization, Regional Office for Europe: Stockholm, Sweden, 2023. [Google Scholar]
- Selvam, A.; Xu, D.; Zhao, Z.; Wong, J.W. Fate of tetracycline, sulfonamide and fluoroquinolone resistance genes and the changes in bacterial diversity during composting of swine manure. Bioresour. Technol. 2012, 126, 383–390. [Google Scholar] [CrossRef]
- Sun, W.; Gu, J.; Wang, X.; Qian, X.; Peng, H. Solid-state anaerobic digestion facilitates the removal of antibiotic resistance genes and mobile genetic elements from cattle manure. Bioresour. Technol. 2019, 274, 287–295. [Google Scholar] [CrossRef]
- Jaya, D.; Ranjeet Kumar, M.; Sampath, C.; Prakash, B.; Naveen, D. A comprehensive study on anaerobic digestion of organic solid waste: A review on configurations, operating parameters, techno-economic analysis and current trends. Biotechnol. Notes 2024, 5, 33–49. [Google Scholar] [CrossRef]
- Borjesson, S.; Mattsson, A.; Lindgren, P.E.; Lindgren, P.E. Genes encoding tetracycline resistance in a full-scale municipal wastewater treatment plant investigated during one year. J. Water Health 2010, 8, 247–256. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.; Ma, Y.; Novak, J.; Pruden, A. Disinfection of Microconstituent Antibiotic Resistance Genes by UV light and Sludge Digestion. Proc. Water Environ. Fed. 2009, 2009, 577–589. [Google Scholar] [CrossRef]
- Ferreira da Silva, M.; Tiago, I.; Verissimo, A.; Boaventura, R.A.; Nunes, O.C.; Manaia, C.M. Antibiotic resistance of enterococci and related bacteria in an urban wastewater treatment plant. FEMS Microbiol. Ecol. 2006, 55, 322–329. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e459. [Google Scholar] [CrossRef]
- Horsman, M.E.; Marous, D.R.; Li, R.; Oliver, R.A.; Byun, B.; Emrich, S.J.; Boggess, B.; Townsend, C.A.; Mobashery, S. Whole-Genome Shotgun Sequencing of Two beta-Proteobacterial Species in Search of the Bulgecin Biosynthetic Cluster. ACS Chem. Biol. 2017, 12, 2552–2557. [Google Scholar] [CrossRef]
- Yap, P.S.; Yiap, B.C.; Ping, H.C.; Lim, S.H. Essential oils, a new horizon in combating bacterial antibiotic resistance. Open Microbiol. J. 2014, 8, 6–14. [Google Scholar] [CrossRef]
- Galgano, M.; Capozza, P.; Pellegrini, F.; Cordisco, M.; Sposato, A.; Sblano, S.; Camero, M.; Lanave, G.; Fracchiolla, G.; Corrente, M.; et al. Antimicrobial Activity of Essential Oils Evaluated In Vitro against Escherichia coli and Staphylococcus aureus. Antibiotics 2022, 11, 979. [Google Scholar] [CrossRef]
- Galgano, M.; Mrenoshki, D.; Pellegrini, F.; Capozzi, L.; Cordisco, M.; Del Sambro, L.; Trotta, A.; Camero, M.; Tempesta, M.; Buonavoglia, D.; et al. Antibacterial and Biofilm Production Inhibition Activity of Thymus vulgaris L. Essential Oil against Salmonella spp. Isolates from Reptiles. Pathogens 2023, 12, 804. [Google Scholar] [CrossRef]
- Galgano, M.; Pellegrini, F.; Mrenoshki, D.; Capozza, P.; Omar, A.; Salvaggiulo, A.; Camero, M.; Lanave, G.; Tempesta, M.; Pratelli, A.; et al. Assessing Contact Time and Concentration of Thymus vulgaris Essential Oil on Antibacterial Efficacy In Vitro. Antibiotics 2023, 12, 1129. [Google Scholar] [CrossRef] [PubMed]
- Iseppi, R.; Mariani, M.; Condo, C.; Sabia, C.; Messi, P. Essential Oils: A Natural Weapon against Antibiotic-Resistant Bacteria Responsible for Nosocomial Infections. Antibiotics 2021, 10, 417. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Antibiotic Targets | Antibiotic Action Mechanism | Antibiotic Class and Major Molecules |
|---|---|---|
| Cell wall | Bactericidal agents limit the formation of peptidoglycan, disrupt peptidoglycan cross-linkage, and impede precursor movement. | Glycopeptide: Vancomycin Bacitracin Beta-lactam: Penicillin, cephalosporin (ceftriaxone, cefotaxime), carbapenem (meropenem) monobactam |
| Cytoplasmic membrane | Bactericidal agents, considered at the time to be the last line of antibiotics in infections caused by MDR pathogens due to their unselective site of action, can cause a toxic effect in humans, such as the nephrotoxic or neurotoxic effect caused by polymyxins, limiting their usability only in restricted dosages. Antibiotics enhance cell permeability, resulting in efflux of cellular contents. | Polymyxins (for Gram-negatives) Gramicidin, Tyrocidine (for Gram-positives) |
| Protein synthesis | Both bactericidal and bacteriostatic agents target the 50s and the 30s ribosomal subunits, inhibiting protein synthesis. | Acting on 50s: Macrolides: Erythromycin Chloramphenicol Oxazolidine Binding site on 50s/Acting in the 30s: Tetracycline Aminoglycosides: Streptomycin, Gentamycin, Neomycin, Kanamycin, Tobramycin, Amikacin |
| Nucleic acid synthesis | Bactericidal antibiotics interfering with bacterial DNA and RNA synthesis | Acting on DNA gyrase: Quinolones: Nalidixic acid Fluoroquinolones: Ciprofloxacin, Norfloxacin, Ofloxacin Aminocoumarin: Novobiocin Acting on RNA polymerase: Rifamycin: Rifampicin |
| Folate synthesis | Bacteriostatic, inhibit the synthesis of DHF * and THF # | Inhibit PABA $ to DHF *: Sulfonamide Inhibit DHF to THF #: Trimethoprim |
| Enzyme Family | Enzyme Types | Reaction Catalyzed | Target Molecules |
|---|---|---|---|
| Acetyltransferases (AAC) | AAC, CAT, VAT | Acetylation | Aminoglycosides, Chloramphenicol, Virginiamycin |
| Phosphotransferase (APH) | APH, CPT | Phosphorylation | Aminoglycosides, Chloramphenicol |
| Adenylyltransferase (ANT) | ANT, LIN | Adenylylation | Aminoglycosides, Lincosamides |
| Macrolide Modifying Enzymes | MPH, Glycosyltransferases, Acetyltransferases | Phosphorylation, Glycosylation, Acetylation | Macrolides, Ketolides, Lincosamides, Streptogramins |
| Fosfomycin Modifying Enzymes | FosA, FosB, FosX, FomA, FomB | Various modifications | Fosfomycin |
| Rifamycin Modifying Enzymes | ADP-ribosyltransferases, Glycosyltransferases, Phosphotransferases, Monooxygenases | Various modifications | Rifamycins |
| Tetracycline Modifying Enzymes | TetX, Rox | Hydroxylation, Oxida tion | Tetracyclines |
| β-lactamases | CTX-M, TEM, SHV, KPC, BLC, OXA, NDM, VIM, IMP | Hydrolysis | β-lactams (Penicillins, Cephalosporins, Carbapenems, Monobactams) |
| Macrolide Esterases (ME) | EreA, EreB | Hydrolysis | Macrolides |
| Mechanism | Description | Examples |
|---|---|---|
| Decreasing Penetration | Limiting the entry of antibiotics into the cell through complex cell walls and porin mutations. | Reduced porin expression, altered porin function |
| Efflux Pumps | Active transport proteins expel antibiotics from the cell using energy from ATP hydrolysis or chemical gradients. | ABC transporters, SMR family, MATE family, MFS, RND family |
| Mechanism | Description | Examples |
|---|---|---|
| Target Protection | Resistance proteins protect the target site from antibiotics. | TetM, TetO (tetracycline resistance), Qnr (quinolone resistance) |
| Point Mutation | Mutations in genes encoding the target site reduce antibiotic affinity. | RNA polymerase (RIF resistance), DNA gyrase (FQ resistance) |
| Enzymatic Alteration | Enzymes modify the target site, reducing antibiotic binding. | Methylation of 23S rRNA (macrolide resistance), Cfr enzyme (linezolid resistance) |
| Target Replacement | Replacement of normal target proteins with resistant variants. | PBP2a (β-lactam resistance) |
| Target Bypass | Increased production of target sites or use of alternative pathways. | Overproduction of DHFR or DHPS (TMP-SMX resistance), use of exogenous folate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galgano, M.; Pellegrini, F.; Catalano, E.; Capozzi, L.; Del Sambro, L.; Sposato, A.; Lucente, M.S.; Vasinioti, V.I.; Catella, C.; Odigie, A.E.; et al. Acquired Bacterial Resistance to Antibiotics and Resistance Genes: From Past to Future. Antibiotics 2025, 14, 222. https://doi.org/10.3390/antibiotics14030222
Galgano M, Pellegrini F, Catalano E, Capozzi L, Del Sambro L, Sposato A, Lucente MS, Vasinioti VI, Catella C, Odigie AE, et al. Acquired Bacterial Resistance to Antibiotics and Resistance Genes: From Past to Future. Antibiotics. 2025; 14(3):222. https://doi.org/10.3390/antibiotics14030222
Chicago/Turabian StyleGalgano, Michela, Francesco Pellegrini, Elisabetta Catalano, Loredana Capozzi, Laura Del Sambro, Alessio Sposato, Maria Stella Lucente, Violetta Iris Vasinioti, Cristiana Catella, Amienwanlen Eugene Odigie, and et al. 2025. "Acquired Bacterial Resistance to Antibiotics and Resistance Genes: From Past to Future" Antibiotics 14, no. 3: 222. https://doi.org/10.3390/antibiotics14030222
APA StyleGalgano, M., Pellegrini, F., Catalano, E., Capozzi, L., Del Sambro, L., Sposato, A., Lucente, M. S., Vasinioti, V. I., Catella, C., Odigie, A. E., Tempesta, M., Pratelli, A., & Capozza, P. (2025). Acquired Bacterial Resistance to Antibiotics and Resistance Genes: From Past to Future. Antibiotics, 14(3), 222. https://doi.org/10.3390/antibiotics14030222