
Genome Analysis of 6222 Bacterial Isolates from Livestock and Food Environments in Spain to Decipher the Antibiotic Resistome

 , , , , , , , , ,
, , , , , , , , ,  , , , and
, , , and 
Abstract
1. Introduction
2. Results
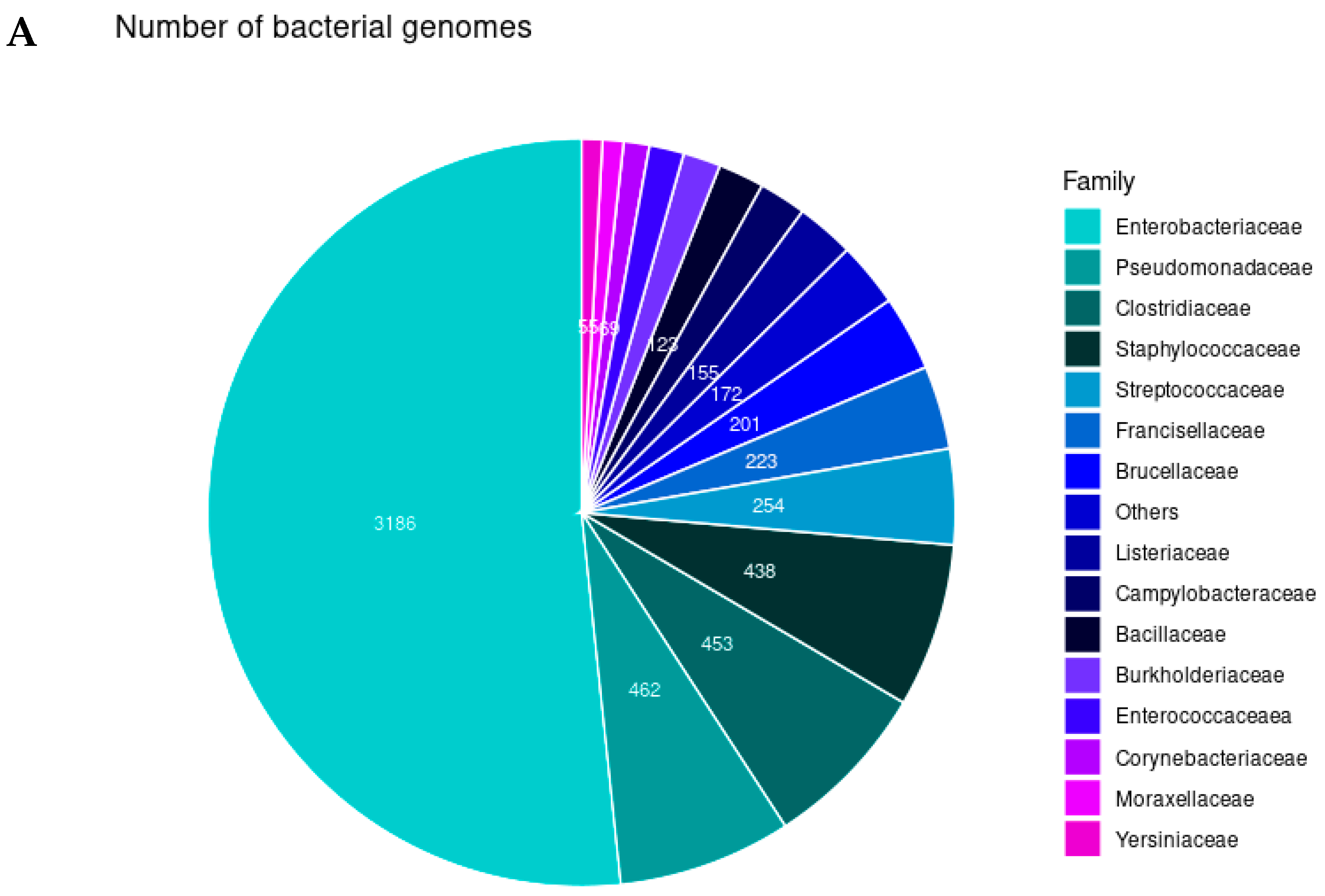
2.1. Bacterial Identification and Gene Distribution
2.2. Plasmids
2.3. Resistome of the Animal Bacterial Isolates in Spain
2.4. Bla Genes
2.5. MDR Correlation of Bacteria
3. Discussion
3.1. Microbial Community Structure
3.2. Antimicrobial Resistance Genes
3.3. Bla Genes
3.4. Linking Antimicrobial Resistance in Livestock to Clinical Cases
4. Materials and Methods
4.1. Samples
4.2. Sequencing
4.3. Accession Numbers
4.4. Genomic Analysis
4.5. Metadata Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burki, T.K. Superbugs: An Arms Race Against Bacteria. Lancet Respir. Med. 2018, 6, 668. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. 2014. Available online: https://wellcomecollection.org/works/rdpck35v (accessed on 3 December 2024).
- Bank, W. Drug-Resistant Infections: A Threat to Our Economic Future. Available online: https://www.worldbank.org/en/topic/health/publication/drug-resistant-infections-a-threat-to-our-economic-future (accessed on 10 September 2024).
- Ahmad, N.; Joji, R.M.; Shahid, M. Evolution and Implementation of One Health to Control the Dissemination of Antibiotic-Resistant Bacteria and Resistance Genes: A Review. Front. Cell Infect. Microbiol. 2022, 12, 1065796. [Google Scholar] [CrossRef]
- Web Annex, A. World Health Organization Model List of Essential Medicines–23rd List, 2023. In The Selection and Use of Essential Medicines 2023: Executive Summary of the Report of the 24th WHO Expert Committee on the Selection and Use of Essential Medicines, 24–28 April 2023; World Health Organization: Geneva, Switzerland, 2023; (WHO/MHP/HPS/EML/2023.02). Licence: CC BY-NC-SA 3.0 IGO; Available online: https://www.who.int/publications/i/item/WHO-MHP-HPS-EML-2023.02 (accessed on 10 September 2024).
- Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions—A Farm to Fork Strategy for a Fair, Healthy and Environmentally-Friendly Food System-COM/2020/381 Final. 2020.
- Chee, M.S.J.; Serrano, E.; Chiang, Y.N.; Harling-Lee, J.; Man, R.; Bacigalupe, R.; Fitzgerald, J.R.; Penadés, J.R.; Chen, J. Dual pathogenicity island transfer by piggybacking lateral transduction. Cell 2023, 186, 3414–3426. [Google Scholar] [CrossRef]
- Tiseo, K.; Huber, L.; Gilbert, M.; Robinson, T.P.; Van Boeckel, T.P. Global Trends in Antimicrobial Use in Food Animals from 2017 to 2030. Antibiotics 2020, 9, 918. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- CDC Antibiotic Resistance Threats Report. Available online: https://www.cdc.gov/antimicrobial-resistance/data-research/threats/index.html (accessed on 15 September 2024).
- Amador, P.; Fernandes, R.; Prudêncio, C.; Duarte, I. Prevalence of Antibiotic Resistance Genes in Multidrug-Resistant Enterobacteriaceae on Portuguese Livestock Manure. Antibiotics 2019, 8, 23. [Google Scholar] [CrossRef]
- Karczmarczyk, M.; Walsh, C.; Slowey, R.; Leonard, N.; Fanning, S. Molecular Characterization of Multidrug-Resistant Escherichia Coli Isolates from Irish Cattle Farms. Appl. Environ. Microbiol. 2011, 77, 7121–7127. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Wang, M.; Zhang, Y.; Fang, C.; Zhang, R.; Fang, L.; Sun, J.; Liu, Y.; Liao, X. Distribution of Antibiotic Resistance Genes and Their Pathogen Hosts in Duck Farm Environments in South-East Coastal China. Appl. Microbiol. Biotechnol. 2024, 108, 136. [Google Scholar] [CrossRef] [PubMed]
- Maciel-Guerra, A.; Baker, M.; Hu, Y.; Wang, W.; Zhang, X.; Rong, J.; Zhang, Y.; Zhang, J.; Kaler, J.; Renney, D.; et al. Dissecting Microbial Communities and Resistomes for Interconnected Humans, Soil, and Livestock. ISME J. 2022, 17, 21–35. [Google Scholar] [CrossRef]
- Gao, F.Z.; He, L.Y.; He, L.X.; Bai, H.; Zhang, M.; Chen, Z.Y.; Qiao, L.K.; Liu, Y.S.; Ying, G.G. Swine Farming Shifted the Gut Antibiotic Resistome of Local People. J. Hazard. Mater. 2024, 465, 133082. [Google Scholar] [CrossRef]
- Lawther, K.; Santos, F.G.; Oyama, L.B.; Rubino, F.; Morrison, S.; Creevey, C.J.; McGrath, J.W.; Huws, S.A. Resistome Analysis of Global Livestock and Soil Microbiomes. Front. Microbiol. 2022, 13, 897905. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Li, H.; Zhou, J.; Wang, T. Seasonal Dissemination of Antibiotic Resistome from Livestock Farms to Surrounding Soil and Air: Bacterial Hosts and Risks for Human Exposure. J. Environ. Manag. 2023, 325, 116638. [Google Scholar] [CrossRef] [PubMed]
- Munk, P.; Yang, D.; Röder, T.; Maier, L.; Petersen, T.N.; Duarte, A.S.R.; Clausen, P.T.L.C.; Brinch, C.; Van Gompel, L.; Luiken, R.; et al. The European Livestock Resistome. mSystems 2024, 9, e0132823. [Google Scholar] [CrossRef]
- Carattoli, A.; Miriagou, V.; Bertini, A.; Loli, A.; Colinon, C.; Villa, L.; Whichard, J.M.; Rossolini, G.M. Replicon Typing of Plasmids Encoding Resistance to Newer β-Lactams. Emerg. Infect. Dis. 2006, 12, 1145. [Google Scholar] [CrossRef] [PubMed]
- Abuoun, M.; Jones, H.; Stubberfield, E.; Gilson, D.; Shaw, L.P.; Hubbard, A.T.M.; Chau, K.K.; Sebra, R.; Peto, T.E.A.; Crook, D.W.; et al. A Genomic Epidemiological Study Shows That Prevalence of Antimicrobial Resistance in Enterobacterales Is Associated with the Livestock Host, as Well as Antimicrobial Usage. Microb. Genom. 2021, 7, 630. [Google Scholar] [CrossRef]
- Baker, M.; Zhang, X.; Maciel-Guerra, A.; Babaarslan, K.; Dong, Y.; Wang, W.; Hu, Y.; Renney, D.; Liu, L.; Li, H.; et al. Convergence of Resistance and Evolutionary Responses in Escherichia coli and Salmonella enterica Co-Inhabiting Chicken Farms in China. Nat. Comm. 2024, 15, 206. [Google Scholar] [CrossRef]
- Villa, L.; García-Fernández, A.; Fortini, D.; Carattoli, A. Replicon Sequence Typing of IncF Plasmids Carrying Virulence and Resistance Determinants. J. Antimicrob. Chemother. 2010, 65, 2518–2529. [Google Scholar] [CrossRef]
- Karslake, J.; Maltas, J.; Brumm, P.; Wood, K.B. Population Density Modulates Drug Inhibition and Gives Rise to Potential Bistability of Treatment Outcomes for Bacterial Infections. PLoS Comput. Biol. 2016, 12, e1005098. [Google Scholar] [CrossRef]
- Tan, C.; Phillip Smith, R.; Srimani, J.K.; Riccione, K.A.; Prasada, S.; Kuehn, M.; You, L. The Inoculum Effect and Band-Pass Bacterial Response to Periodic Antibiotic Treatment. Mol. Syst. Biol. 2012, 8, 617. [Google Scholar] [CrossRef]
- Aranda-Díaz, A.; Obadia, B.; Dodge, R.; Thomsen, T.; Hallberg, Z.F.; Güvener, Z.T.; Ludington, W.B.; Huang, K.C. Bacterial Interspecies Interactions Modulate PH-Mediated Antibiotic Tolerance. Elife 2020, 9, e51493. [Google Scholar] [CrossRef]
- Ghuneim, L.A.J.; Raghuvanshi, R.; Neugebauer, K.A.; Guzior, D.V.; Christian, M.H.; Schena, B.; Feiner, J.M.; Castillo-Bahena, A.; Mielke, J.; McClelland, M.; et al. Complex and Unexpected Outcomes of Antibiotic Therapy against a Polymicrobial Infection. ISME J. 2022, 16, 2065–2075. [Google Scholar] [CrossRef] [PubMed]
- Denk-Lobnig, M.; Wood, K.B. Antibiotic Resistance in Bacterial Communities. Curr. Opin. Microbiol. 2023, 74, 102306. [Google Scholar] [CrossRef] [PubMed]
- Sorg, R.A.; Lin, L.; van Doorn, G.S.; Sorg, M.; Olson, J.; Nizet, V.; Veening, J.W. Collective Resistance in Microbial Communities by Intracellular Antibiotic Deactivation. PLoS Biol. 2016, 14, e2000631. [Google Scholar] [CrossRef]
- Nicoloff, H.H.; Andersson, D.I. Indirect Resistance to Several Classes of Antibiotics in Cocultures with Resistant Bacteria Expressing Antibiotic-Modifying or -Degrading Enzymes. J. Antimicrob. Chem. 2016, 71, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Langevin, A.M.; Dunlop, M.J. Antibiotic Export by Efflux Pumps Affects Growth of Neighboring Bacteria. Sci. Rep. 2018, 8, 15120. [Google Scholar] [CrossRef]
- Bottery, M.J.; Wood, A.J.; Brockhurst, M.A. Selective Conditions for a Multidrug Resistance Plasmid Depend on the Sociality of Antibiotic Resistance. Antimicrob. Agents Chemother. 2016, 60, 2524–2527. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Indugu, N.; Toth, J.D.; Bender, J.S.; Baker, L.D.; Hennessy, M.L.; Vecchiarelli, B.; Aceto, H.; Dou, Z. The Distribution of Microbiomes and Resistomes across Farm Environments in Conventional and Organic Dairy Herds in Pennsylvania. Environ. Microbiomes 2020, 15, 21. [Google Scholar] [CrossRef]
- He, Y.; Yuan, Q.; Mathieu, J.; Stadler, L.; Senehi, N.; Sun, R.; Alvarez, P.J.J. Antibiotic Resistance Genes from Livestock Waste: Occurrence, Dissemination, and Treatment. npj Clean Water 2020, 3, 4. [Google Scholar] [CrossRef]
- Munk, P.; Knudsen, B.E.; Lukjacenko, O.; Duarte, A.S.R.; Van Gompel, L.; Luiken, R.E.C.; Smit, L.A.M.; Schmitt, H.; Garcia, A.D.; Hansen, R.B.; et al. Abundance and Diversity of the Faecal Resistome in Slaughter Pigs and Broilers in Nine European Countries. Nat. Microbiol. 2018, 3, 898–908. [Google Scholar] [CrossRef]
- Xiao, L.; Estellé, J.; Kiilerich, P.; Ramayo-Caldas, Y.; Xia, Z.; Feng, Q.; Liang, S.; Pedersen, A.; Kjeldsen, N.J.; Liu, C.; et al. A Reference Gene Catalogue of the Pig Gut Microbiome. Nat. Microbiol. 2016, 1, 16161. [Google Scholar] [CrossRef]
- Joyce, A.; McCarthy, C.G.P.; Murphy, S.; Walsh, F. Antibiotic Resistomes of Healthy Pig Faecal Metagenomes. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tian, X.; Han, B.; Zhao, R.; Li, J.; Zhang, K. Tracking High-Risk β-Lactamase Gene (Bla Gene) Transfers in Two Chinese Intensive Dairy Farms. Environ. Poll. 2021, 274, 116593. [Google Scholar] [CrossRef]
- Kim, J.; Cho, Y.; Lim, S.K.; Seo, M.R.; Sohn, J.W.; Kim, B.; Rho, M.; Pai, H. Comparative Analyses of the Faecal Resistome against β-Lactam and Quinolone Antibiotics in Humans and Livestock Using Metagenomic Sequencing. Sci. Rep. 2023, 13, 20993. [Google Scholar] [CrossRef] [PubMed]
- WHO. One Health. Available online: https://www.who.int/health-topics/one-health#tab=tab_1 (accessed on 11 September 2024).
- Peng, Z.; Maciel-Guerra, A.; Baker, M.; Zhang, X.; Hu, Y.; Wang, W.; Rong, J.; Zhang, J.; Xue, N.; Barrow, P.; et al. Whole-Genome Sequencing and Gene Sharing Network Analysis Powered by Machine Learning Identifies Antibiotic Resistance Sharing between Animals, Humans and Environment in Livestock Farming. PLoS Comput. Biol. 2022, 18, e1010018. [Google Scholar] [CrossRef]
- Sun, J.; Liao, X.P.; D’Souza, A.W.; Boolchandani, M.; Li, S.H.; Cheng, K.; Luis Martínez, J.; Li, L.; Feng, Y.J.; Fang, L.X.; et al. Environmental Remodeling of Human Gut Microbiota and Antibiotic Resistome in Livestock Farms. Nat. Comm. 2020, 11, 1427. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, X.; Li, J.; Lv, N.; Liu, F.; Wu, J.; Lin, I.Y.C.; Wu, N.; Weimer, B.C.; Gao, G.F.; et al. The Bacterial Mobile Resistome Transfer Network Connecting the Animal and Human Microbiomes. Appl. Environ. Microbiol. 2016, 82, 6672–6681. [Google Scholar] [CrossRef]
- Wang, Y.; Lyu, N.; Liu, F.; Liu, W.J.; Bi, Y.; Zhang, Z.; Ma, S.; Cao, J.; Song, X.; Wang, A.; et al. More Diversified Antibiotic Resistance Genes in Chickens and Workers of the Live Poultry Markets. Environ. Int. 2021, 153, 106534. [Google Scholar] [CrossRef]
- Kim, J.; Cho, Y.; Seo, M.R.; Bae, M.H.; Kim, B.; Rho, M.; Pai, H. Quantitative Characterization of Clostridioides Difficile Population in the Gut Microbiome of Patients with C. difficile Infection and Their Association with Clinical Factors. Sci. Rep. 2020, 10, 17608. [Google Scholar] [CrossRef] [PubMed]
- Pustam, A.; Jayaraman, J.; Ramsubhag, A. Whole Genome Sequencing Reveals Complex Resistome Features of Klebsiella Pneumoniae Isolated from Patients at Major Hospitals in Trinidad, West Indies. J. Glob. Antimicrob. Resist. 2024, 37, 141–149. [Google Scholar] [CrossRef]
- Filippa, N.; Carricajo, A.; Grattard, F.; Fascia, P.; Sayed, F.E.; Defilippis, J.P.; Berthelot, P.; Aubert, G. Outbreak of Multidrug-Resistant Klebsiella pneumoniae Carrying QnrB1 and BlaCTX-M15 in a French Intensive Care Unit. Ann. Intensive Care 2013, 3, 18. [Google Scholar] [CrossRef]
- Rahmat Ullah, S.; Irum, S.; Mahnoor, I.; Ismatullah, H.; Mumtaz, M.; Andleeb, S.; Rahman, A.; Jamal, M. Exploring the Resistome, Virulome, and Mobilome of Multidrug-Resistant Klebsiella pneumoniae Isolates: Deciphering the Molecular Basis of Carbapenem Resistance. BMC Genom. 2024, 25, 408. [Google Scholar] [CrossRef] [PubMed]
- Mbelle, N.M.; Feldman, C.; Sekyere, J.O.; Maningi, N.E.; Modipane, L.; Essack, S.Y. Pathogenomics and Evolutionary Epidemiology of Multi-Drug Resistant Clinical Klebsiella pneumoniae Isolated from Pretoria, South Africa. Sci. Rep. 2020, 10, 1232. [Google Scholar] [CrossRef]
- Founou, R.C.; Founou, L.L.; Allam, M.; Ismail, A.; Essack, S.Y. Whole Genome Sequencing of Extended Spectrum β-Lactamase (ESBL)-Producing Klebsiella pneumoniae Isolated from Hospitalized Patients in KwaZulu-Natal, South Africa. Sci. Rep. 2019, 9, 6266. [Google Scholar] [CrossRef]
- Quijada, N.M.; Rodríguez-Lázaro, D.; Eiros, J.M.; Hernández, M. TORMES: An Automated Pipeline for Whole Bacterial Genome Analysis. Bioinformatics 2019, 35, 4207–4212. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality Control and Preprocessing of Metagenomic Datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Seemann, T. Abricate, Github. Available online: https://Github.Com/Tseemann/Abricate (accessed on 3 December 2024).
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A Reference Database for Bacterial Virulence Factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Taxa | Number of Isolates |
|---|---|
| Family | |
| Acetobacteraceae | 1 |
| Brucellaceae | 201 |
| Burkholderiaceae | 100 |
| Caulobacteraceae | 1 |
| Francisellaceae | 223 |
| Leuconostocaceae | 2 |
| Mycoplasmataceae | 12 |
| Nocardiaceae | 1 |
| Paenibacillaceae | 6 |
| Peptostreptococcaceae | 126 |
| Species | |
| Acetobacter ghanensis | 1 |
| Aerococcus sanguinicola | 1 |
| Aerococcus urinae | 1 |
| Aerococcus urinaeequi | 3 |
| Aerococcus urinaehominis | 1 |
| Aerococcus viridans | 1 |
| Aeromonas veronii | 1 |
| Anoxybacillus flavithermus | 4 |
| Bacillus amyloliquefaciens | 1 |
| Bacillus atrophaeus | 1 |
| Bacillus cereus | 50 |
| Bacillus megaterium | 1 |
| Bacillus velezensis | 1 |
| Lysinibacillus sp. | 1 |
| Brucella abortus | 129 |
| Brucella ceti | 17 |
| Brucella melitensis | 35 |
| Brucella pinnipedialis | 2 |
| Brucella sp. | 1 |
| Brucella suis | 17 |
| Burkholderia contaminans | 12 |
| Burkholderia multivorans | 37 |
| Burkholderia vietnamiensis | 2 |
| Cupriavidus metallidurans | 2 |
| Ralstonia mannitolilytica | 27 |
| Ralstonia pickettii | 20 |
| Brevundimonas diminuta | 1 |
| Clostridium septicum | 8 |
| Clostridium sphenoides | 1 |
| Clostridium tetani | 3 |
| Delftia tsuruhatensis | 1 |
| Corynebacterium striatum | 1 |
| Corynebacterium ulcerans | 4 |
| Corynebacterium variabile | 1 |
| Atlantibacter hermannii | 2 |
| Pantoea alhagi | 3 |
| Francisella tularensis | 223 |
| Lactobacillus curvatus | 2 |
| Lactobacillus mali | 4 |
| Lactobacillus plantarum | 1 |
| Acinetobacter lwoffii | 1 |
| Providencia alcalifaciens | 2 |
| Mycoplasma orale | 12 |
| Rhodococcus aetherivorans | 1 |
| Brevibacillus laterosporus | 2 |
| Paenibacillus cellulositrophicus | 1 |
| Paenibacillus polymyxa | 2 |
| Paenibacillus xylanexedens | 1 |
| Pasteurella multocida | 7 |
| Paeniclostridium sordellii | 5 |
| Pseudomonas fluorescens | 1 |
| Pseudomonas koreensis | 1 |
| Pseudomonas protegens | 3 |
| Rahnella aquatilis | 1 |
| Serratia fonticola | 1 |
| Serratia proteamaculans | 1 |
| Family | Species | Number of Resistance Genes | % Strains |
|---|---|---|---|
| Enterobacteriaceae | Escherichia coli | 1680 | 32.93% |
| Klebsiella pneumoniae | 390 | 6.30% | |
| Salmonella enterica | 281 | 9.23% | |
| Citrobacter freundii | 21 | 0.40% | |
| Enterobacter cloacae | 25 | 0.40% | |
| Enterobacter hormaechei | 38 | 0.74% | |
| Pseudomonadaceae | Pseudomonas aeruginosa | 401 | 6.44% |
| Pseudomonas putida | 30 | 0.56% | |
| Moraxellaceae | Acinetobacter baumannii | 54 | 0.87% |
| Staphylococcaceae | Staphylococcus aureus | 296 | 5.79% |
| Clostridiaceae | Clostridium perfringens | 209 | 4.60% |
| Streptococcaceae | Streptococcus suis | 40 | 0.84% |
| Lactococcus garvieae | 150 | 3.57% | |
| Enterococcaceae | Enterococcus faecium | 50 | 0.80% |
| Campylobacteraceae | Campylobacter coli | 70 | 1.74% |
| Campylobacter jejuni | 29 | 0.69% | |
| Peptostreptococcaceae | Clostridioides difficile | 47 | 0.76% |
| Yersiniaceae | Yersinia enterocolitica | 28 | 0.67% |
| Morganellaceae | Morganella morganii | 10 | 0.16% |
| Providencia rettgeri | 6 | 0.14% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, M.; Falcó-Prieto, Á.; Ugarte-Ruiz, M.; Miguela-Villoldo, P.; Ocampo-Sosa, A.; Abad, D.; Pérez-Sancho, M.; Álvarez, J.; Cadamuro, R.D.; Elois, M.A.; et al. Genome Analysis of 6222 Bacterial Isolates from Livestock and Food Environments in Spain to Decipher the Antibiotic Resistome. Antibiotics 2025, 14, 281. https://doi.org/10.3390/antibiotics14030281
Hernández M, Falcó-Prieto Á, Ugarte-Ruiz M, Miguela-Villoldo P, Ocampo-Sosa A, Abad D, Pérez-Sancho M, Álvarez J, Cadamuro RD, Elois MA, et al. Genome Analysis of 6222 Bacterial Isolates from Livestock and Food Environments in Spain to Decipher the Antibiotic Resistome. Antibiotics. 2025; 14(3):281. https://doi.org/10.3390/antibiotics14030281
Chicago/Turabian StyleHernández, Marta, Álvaro Falcó-Prieto, Maria Ugarte-Ruiz, Pedro Miguela-Villoldo, Alain Ocampo-Sosa, David Abad, Marta Pérez-Sancho, Julio Álvarez, Rafael Dorighello Cadamuro, Mariana Alves Elois, and et al. 2025. "Genome Analysis of 6222 Bacterial Isolates from Livestock and Food Environments in Spain to Decipher the Antibiotic Resistome" Antibiotics 14, no. 3: 281. https://doi.org/10.3390/antibiotics14030281
APA StyleHernández, M., Falcó-Prieto, Á., Ugarte-Ruiz, M., Miguela-Villoldo, P., Ocampo-Sosa, A., Abad, D., Pérez-Sancho, M., Álvarez, J., Cadamuro, R. D., Elois, M. A., Fongaro, G., Quesada, A., González-Zorn, B., Domínguez, L., Eiros, J. M., & Rodríguez-Lázaro, D. (2025). Genome Analysis of 6222 Bacterial Isolates from Livestock and Food Environments in Spain to Decipher the Antibiotic Resistome. Antibiotics, 14(3), 281. https://doi.org/10.3390/antibiotics14030281