
A One Health Approach Metagenomic Study on Antimicrobial Resistance Traits of Canine Saliva
 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results
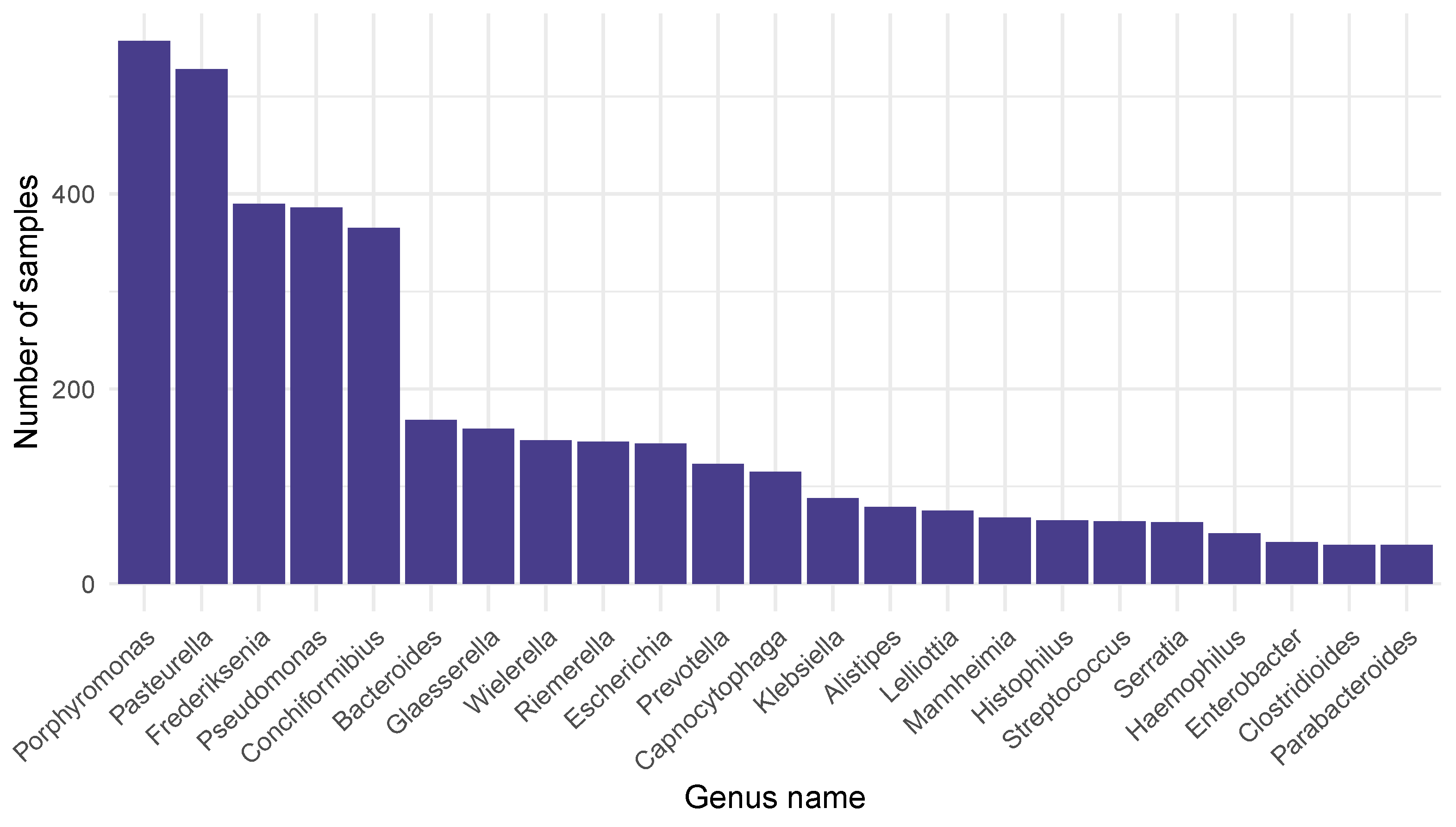
2.1. Bacteriome
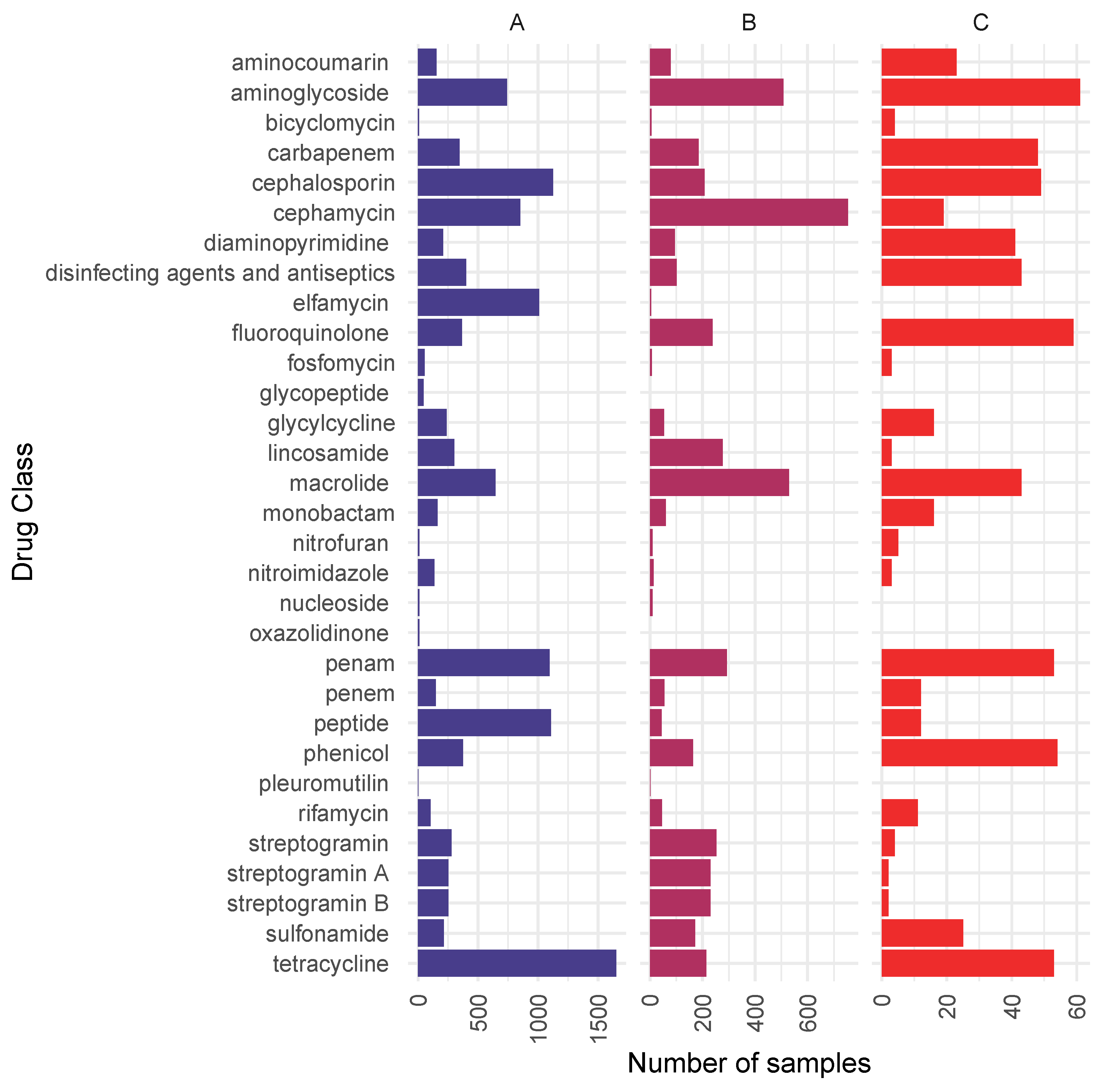
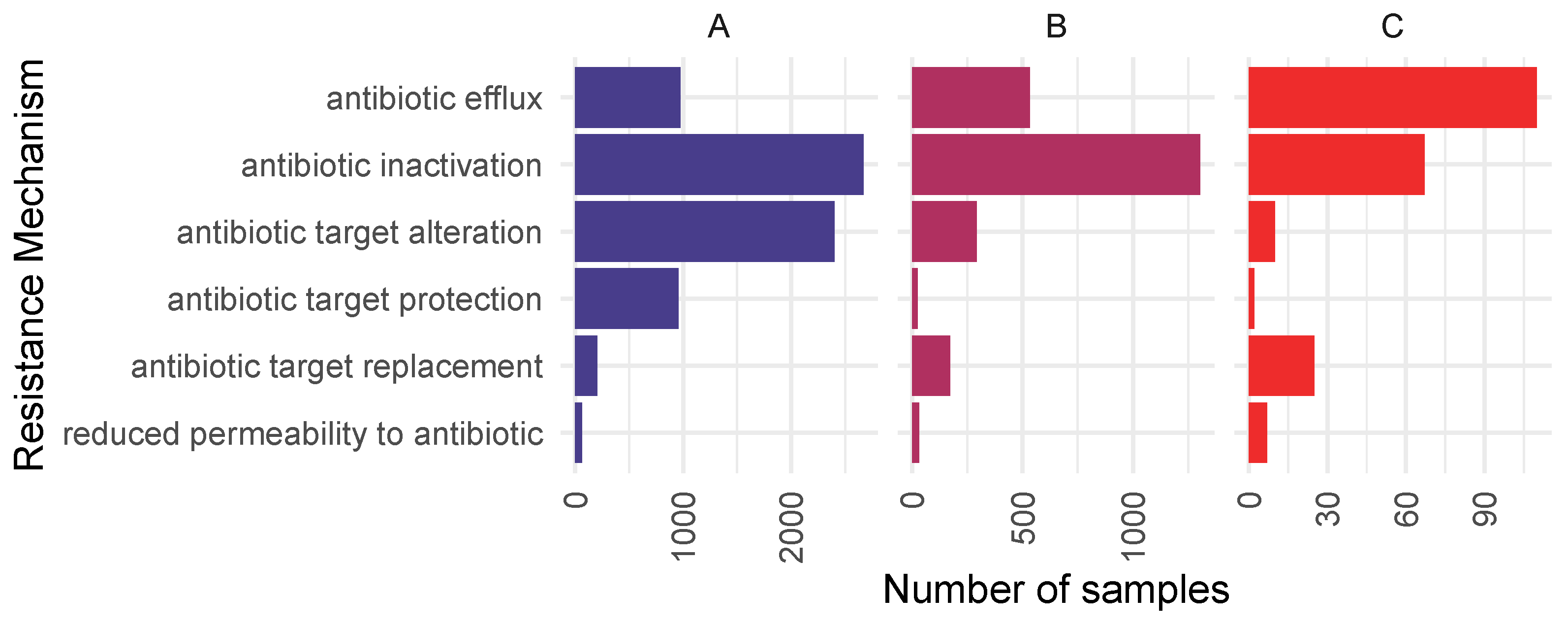
2.2. Resistome
2.3. Basic Characteristics, Physical Traits, and Behavioral Characteristics Associated with ARGs
2.3.1. Basic Characteristics and Physical Traits
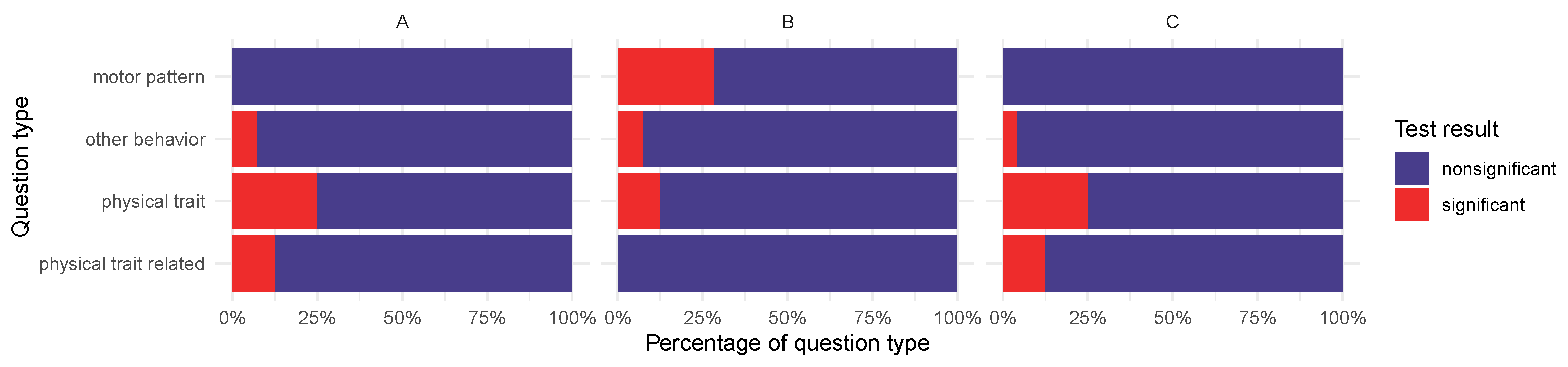
2.3.2. Behavioral Traits
3. Discussion
4. Materials and Methods
4.1. Data
4.2. Bioinformatic and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rowan, A.N. Companion Animal Statistics in the USA. Demography and Statistics for Companion Animal Populations Collection 2018. 7. Available online: https://www.wellbeingintlstudiesrepository.org/demscapop/7 (accessed on 14 April 2025).
- Bedford, E. Number of Dogs in the United States from 2000 to 2017; Statista; American Pet Products Association: Greenwhich, WC, USA, 2019; Available online: https://www.statista.com/statistics/198100/dogs-in-the-united-states-since-2000/#:~:text=According%20to%20a%20pet%20owners,Why%20has%20this%20figure%20increased%3F (accessed on 14 April 2025).
- American Pet Products Association. 2021–2022 APPA National Pet Owners Survey; American Pet Products Association: Greenwhich, WC, USA, 2021. [Google Scholar]
- Jensen, H. AVMA Pet Ownership and Demographics Sourcebook: 2017–2018 Edition; American Veterinary Medical Association: Schaumburg, IL, USA, 2017; ISBN 978-1-882691-52. [Google Scholar]
- Overall, K.L.; Love, M. Dog bites to humans—Demography, epidemiology, injury, and risk. J. Am. Vet. Med. Assoc. 2001, 218, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Q.; Wang, T.; Xu, N.; Lu, T.; Hong, W.; Penuelas, J.; Gillings, M.; Wang, M.; Gao, W.; et al. Assessment of global health risk of antibiotic resistance genes. Nat. Commun. 2022, 13, 1553. [Google Scholar] [CrossRef] [PubMed]
- Morrill, K.; Hekman, J.; Li, X.; McClure, J.; Logan, B.; Goodman, L.; Gao, M.; Dong, Y.; Alonso, M.; Carmichael, E.; et al. Ancestry-inclusive dog genomics challenges popular breed stereotypes. Science 2022, 376, eabk0639. [Google Scholar] [CrossRef] [PubMed]
- Portilho, F.V.; Nóbrega, J.; de Almeida, B.O.; Mota, A.R.; de Paula, C.L.; Listoni, F.J.; Bosco, S.M.; Oliveira, A.L.; Cunha, M.d.L.R.; Ribeiro, M.G. Microbial Complexity of Oral Cavity of Healthy Dogs Identified by Mass Spectrometry and Next-Generation Sequencing. Animals 2023, 13, 2467. [Google Scholar] [CrossRef]
- Mei, S.; Cai, M.; Lei, F.; Wang, X.; Yuan, X.; Lin, Y.; Zhu, B. Revealing microbial community characteristics in healthy human, cat and canine salivas and looking for species-specific microbes. Int. J. Leg. Med. 2024, 138, 2259–2269. [Google Scholar] [CrossRef]
- Alessandri, G.; Fontana, F.; Mancabelli, L.; Tarracchini, C.; Lugli, G.A.; Argentini, C.; Longhi, G.; Rizzo, S.M.; Vergna, L.M.; Anzalone, R.; et al. Species-level characterization of saliva and dental plaque microbiota reveals putative bacterial and functional biomarkers of periodontal diseases in dogs. FEMS Microbiol. Ecol. 2024, 100, fiae082. [Google Scholar] [CrossRef]
- McArthur, A.G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad, M.A.; Baylay, A.J.; Bhullar, K.; Canova, M.J.; De Pascale, G.; Ejim, L.; et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
- Poole, K.; Krebes, K.; McNally, C.; Neshat, S. Multiple antibiotic resistance in Pseudomonas aeruginosa: Evidence for involvement of an efflux operon. J. Bacteriol. 1993, 175, 7363–7372. [Google Scholar] [CrossRef]
- World Health Organization. Critically Important Antimicrobials for Human Medicine; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Tompson, A.C.; Mateus, A.L.; Brodbelt, D.C.; Chandler, C.I. Understanding antibiotic use in companion animals: A literature review identifying avenues for future efforts. Front. Vet. Sci. 2021, 8, 719547. [Google Scholar] [CrossRef]
- Yushchuk, O.; Binda, E.; Marinelli, F. Glycopeptide antibiotic resistance genes: Distribution and function in the producer actinomycetes. Front. Microbiol. 2020, 11, 1173. [Google Scholar] [CrossRef]
- El-Sayed Ahmed, M.A.E.G.; Zhong, L.L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.B. Colistin and its role in the Era of antibiotic resistance: An extended review (2000–2019). Emerg. Microbes Infect. 2020, 9, 868–885. [Google Scholar] [CrossRef]
- Moffatt, J.H.; Harper, M.; Boyce, J.D. Mechanisms of polymyxin resistance. In Polymyxin Antibiotics: From Laboratory Bench to Bedside; Springer: Cham, Switzerland, 2019; pp. 55–71. [Google Scholar]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [PubMed]
- Scientific Advisory Group on Antimicrobials of the Committee for Medicinal Products for Veterinary Use. Reflection paper on the use of third and fourth generation cephalosporins in food producing animals in the European Union: Development of resistance and impact on human and animal health. J. Vet. Pharmacol. Ther. 2009, 32, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Taniguchi, K.; Urasawa, S. Analysis of diversity of mutations in the mecI gene and mecA promoter/operator region of methicillin-resistant Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob. Agents Chemother. 1998, 42, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: A review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]
- O’Neill, D.; Pegram, C.; Crocker, P.; Brodbelt, D.; Church, D.; Packer, R. Unravelling the health status of brachycephalic dogs in the UK using multivariable analysis. Sci. Rep. 2020, 10, 17251. [Google Scholar] [CrossRef]
- Teng, K.T.y.; Brodbelt, D.C.; Pegram, C.; Church, D.B.; O’Neill, D.G. Life tables of annual life expectancy and mortality for companion dogs in the United Kingdom. Sci. Rep. 2022, 12, 6415. [Google Scholar] [CrossRef]
- Bellumori, T.P.; Famula, T.R.; Bannasch, D.L.; Belanger, J.M.; Oberbauer, A.M. Prevalence of inherited disorders among mixed-breed and purebred dogs: 27,254 cases (1995–2010). J. Am. Vet. Med. Assoc. 2013, 242, 1549–1555. [Google Scholar] [CrossRef]
- Singleton, D.A.; Pinchbeck, G.L.; Radford, A.D.; Arsevska, E.; Dawson, S.; Jones, P.H.; Noble, P.J.M.; Williams, N.J.; Sánchez-Vizcaíno, F. Factors associated with prescription of antimicrobial drugs for dogs and cats, United Kingdom, 2014–2016. Emerg. Infect. Dis. 2020, 26, 1778. [Google Scholar] [CrossRef]
- Karlsson, E.K.; Baranowska, I.; Wade, C.M.; Salmon Hillbertz, N.H.; Zody, M.C.; Anderson, N.; Biagi, T.M.; Patterson, N.; Pielberg, G.R.; Kulbokas, E.J., III; et al. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat. Genet. 2007, 39, 1321–1328. [Google Scholar] [CrossRef]
- Deane-Coe, P.E.; Chu, E.T.; Slavney, A.; Boyko, A.R.; Sams, A.J. Direct-to-consumer DNA testing of 6,000 dogs reveals 98.6-kb duplication associated with blue eyes and heterochromia in Siberian Huskies. PLoS Genet. 2018, 14, e1007648. [Google Scholar] [CrossRef]
- Roberts, S.R. Color dilution and hereditary defects in collie dogs. Am. J. Ophthalmol. 1967, 63, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.A.; Wahl, J.M.; Rees, C.A.; Strain, G.M.; Cargill, E.J.; Vanderlip, S.L.; Murphy, K.E. Canine SINEs and their effects on phenotypes of the domestic dog. In Genomics of Disease; Springer: Berlin/Heidelberg, Germany, 2008; pp. 79–88. [Google Scholar]
- Stritzel, S.; Fritsche, J.; Wöhlke, A.; Philipp, U.; Hertslet, S.; Distl, O. Multiple ocular malformations in two sheepdogs homozygous for the merle mutation. Tierarztl. Prax. Ausg. K Kleintiere/Heimtiere 2009, 37, 229–238. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Lahti, L.; Shetty, S. Microbiome R Package. 2012–2019. Available online: https://microbiome.github.io/microbiome (accessed on 14 April 2025).
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2016, 45, gkw1004. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Agresti, A. Categorical Data Analysis; John Wiley & Sons: Hoboken, NJ, USA, 2019; p. 46. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Approach | Number of Samples | OR (95%CI) | p-Value | |||
|---|---|---|---|---|---|---|---|
| Group 1/Group 2 | Group 1 | Group 2 | |||||
| ARG+ | ARG− | ARG+ | ARG− | ||||
| Brachycephal/dolio-, mesocephal | A | 71 | 12 | 606 | 50 | 0.49 (0.24–1.06) | 0.055 |
| B | 44 | 39 | 370 | 286 | 0.87 (0.54–1.42) | 0.560 | |
| C | 7 | 105 | 29 | 598 | 1.37 (0.50–3.31) | 0.473 | |
| Breeder/rescue, shelter | A | 286 | 28 | 1013 | 86 | 0.87 (0.55–1.41) | 0.557 |
| B | 172 | 142 | 628 | 472 | 0.91 (0.70–1.18) | 0.478 | |
| C | 13 | 301 | 33 | 1067 | 1.40 (0.67–2.77) | 0.366 | |
| Females/males | A | 827 | 68 | 852 | 83 | 1.18 (0.84–1.68) | 0.350 |
| B | 511 | 384 | 519 | 416 | 1.13 (0.76–1.70) | 0.561 | |
| C | 27 | 828 | 33 | 861 | 0.85 (0.49–1.47) | 0.600 | |
| Intact/sterilised | A | 87 | 10 | 1515 | 131 | 0.75 (0.38–1.66) | 0.440 |
| B | 44 | 53 | 938 | 709 | 0.63 (0.41–0.97) | 0.027 * | |
| C | 2 | 95 | 58 | 1589 | 0.58 (0.07–2.24) | 0.771 | |
| Purebred/mutt | A | 677 | 62 | 935 | 80 | 0.93 (0.65–1.34) | 0.723 |
| B | 414 | 325 | 577 | 439 | 0.97 (0.80–1.18) | 0.770 | |
| C | 36 | 703 | 25 | 991 | 2.03 (1.17–3.56) | 0.008 ** | |
| Rural, suburban/urban | A | 575 | 46 | 837 | 76 | 1.13 (0.76–1.70) | 0.564 |
| B | 346 | 275 | 515 | 399 | 0.97 (0.79–1.20) | 0.834 | |
| C | 23 | 598 | 31 | 883 | 1.10 (0.60–1.96) | 0.779 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, A.G.; Tóth, D.L.; Remport, L.; Tóth, I.; Németh, T.; Dubecz, A.; Patai, Á.V.; Wagenhoffer, Z.; Makrai, L.; Solymosi, N. A One Health Approach Metagenomic Study on Antimicrobial Resistance Traits of Canine Saliva. Antibiotics 2025, 14, 433. https://doi.org/10.3390/antibiotics14050433
Tóth AG, Tóth DL, Remport L, Tóth I, Németh T, Dubecz A, Patai ÁV, Wagenhoffer Z, Makrai L, Solymosi N. A One Health Approach Metagenomic Study on Antimicrobial Resistance Traits of Canine Saliva. Antibiotics. 2025; 14(5):433. https://doi.org/10.3390/antibiotics14050433
Chicago/Turabian StyleTóth, Adrienn Gréta, Darinka Lilla Tóth, Laura Remport, Imre Tóth, Tibor Németh, Attila Dubecz, Árpád V. Patai, Zsombor Wagenhoffer, László Makrai, and Norbert Solymosi. 2025. "A One Health Approach Metagenomic Study on Antimicrobial Resistance Traits of Canine Saliva" Antibiotics 14, no. 5: 433. https://doi.org/10.3390/antibiotics14050433
APA StyleTóth, A. G., Tóth, D. L., Remport, L., Tóth, I., Németh, T., Dubecz, A., Patai, Á. V., Wagenhoffer, Z., Makrai, L., & Solymosi, N. (2025). A One Health Approach Metagenomic Study on Antimicrobial Resistance Traits of Canine Saliva. Antibiotics, 14(5), 433. https://doi.org/10.3390/antibiotics14050433