
Distribution of Antimicrobial Resistance and Biofilm Production Genes in the Genomic Sequences of S. aureus: A Global In Silico Analysis

 ,
,  ,
,  , , and
, , and 
Abstract
1. Introduction
1.1. Resistance and Dissemination Potential
1.2. Biofilm Architecture and Composition Variability
1.3. Genes Associated with Antimicrobial Resistance and Mobile Genetic Elements
1.4. Whole Genome Sequencing (WGS) Technology and the Formation of Big Data
1.5. The One Health Concept and the Evolution of S. aureus
2. Results
2.1. Analysis of S. aureus Genomes: Geographic Distribution, Clonal Complexes, and SCCmec Isotypes
2.2. Biofilm Genes
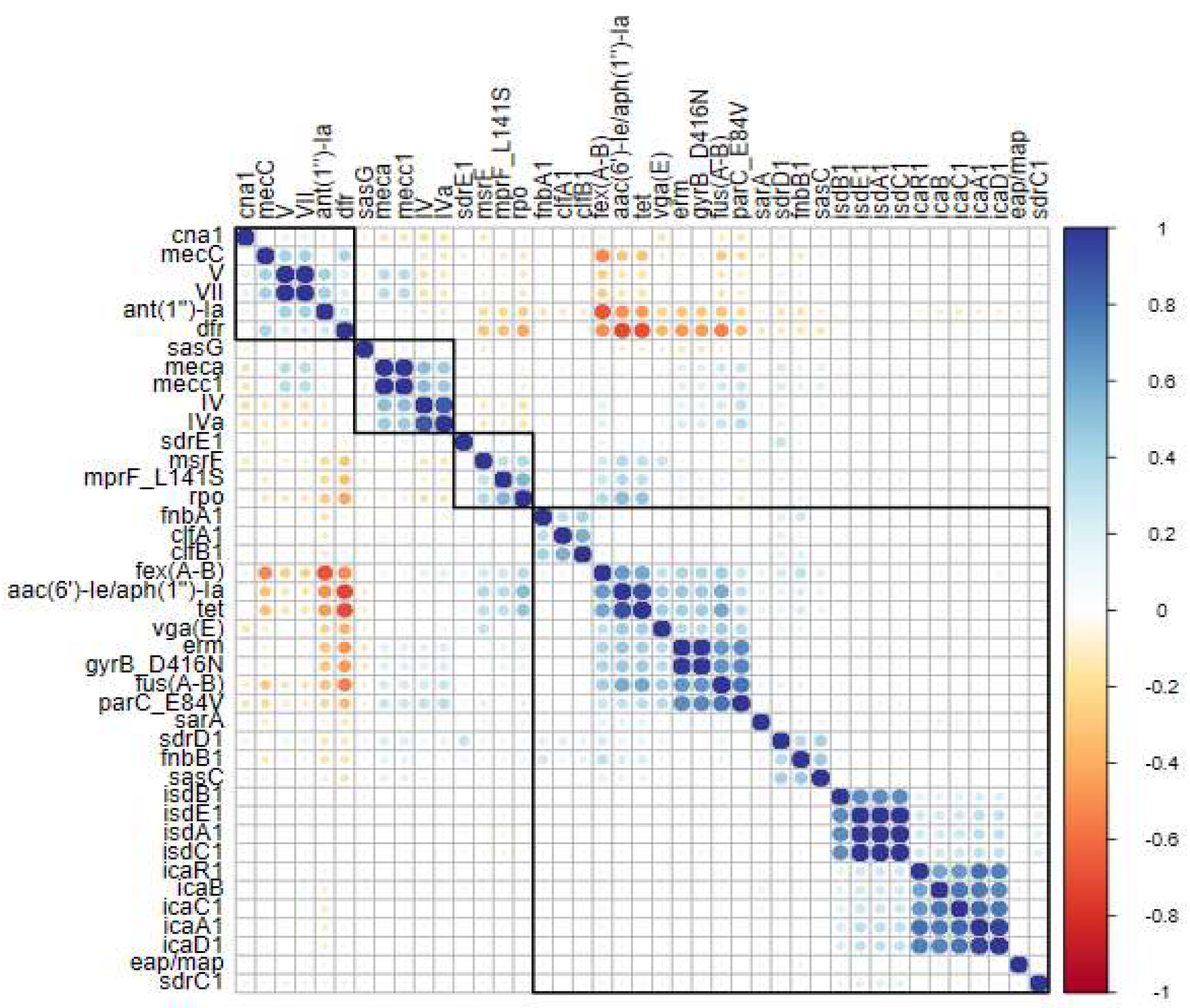
2.3. Correlation Between Resistance Genes and Biofilm-Associated Genes
2.4. Gene Clustering and Correlations in Human Clinical Sequences
2.5. Gene Clustering and Correlations in Environmental Sequences
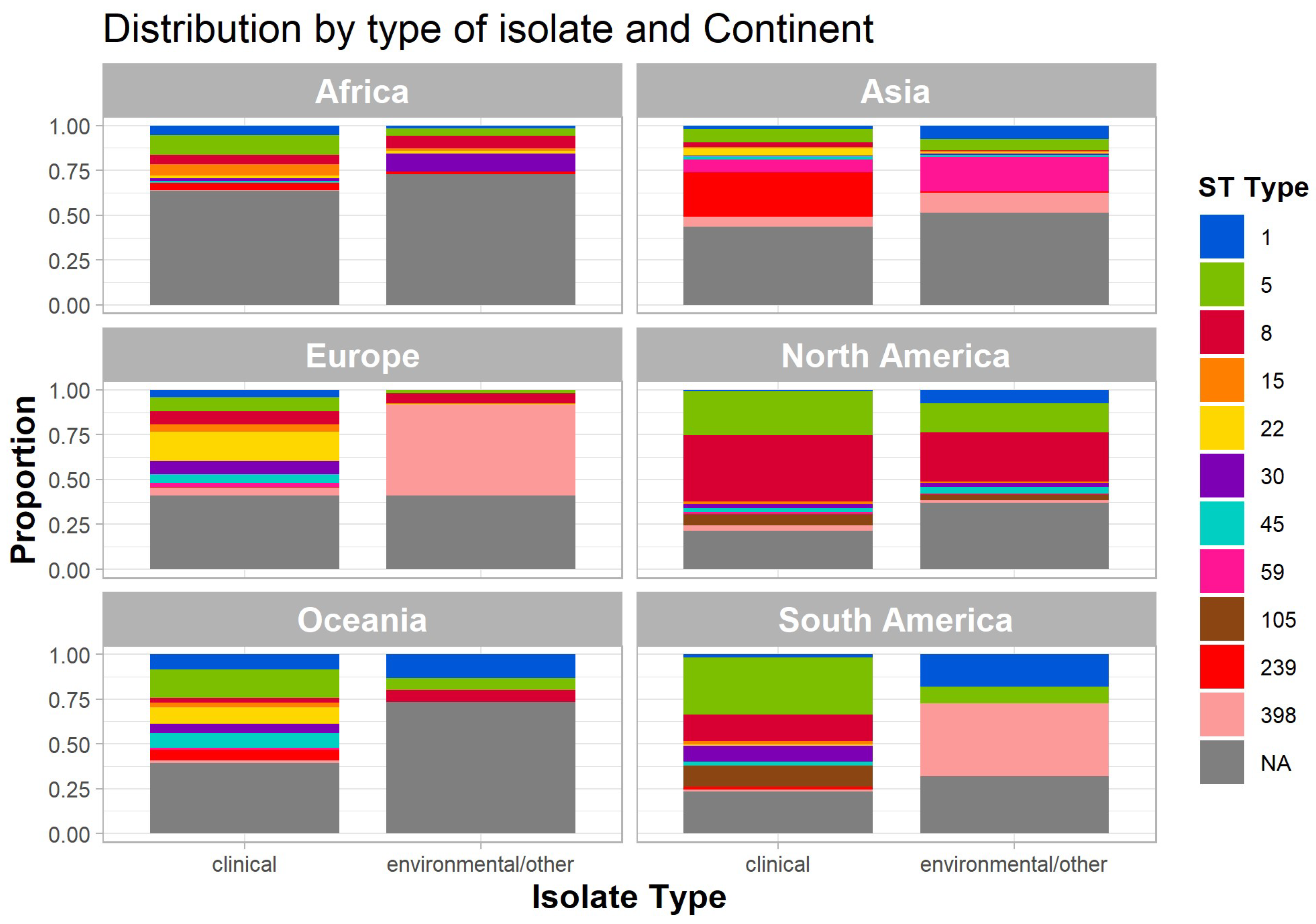
2.6. Global Distribution of MLST in Human Clinical and Environmental Sequences
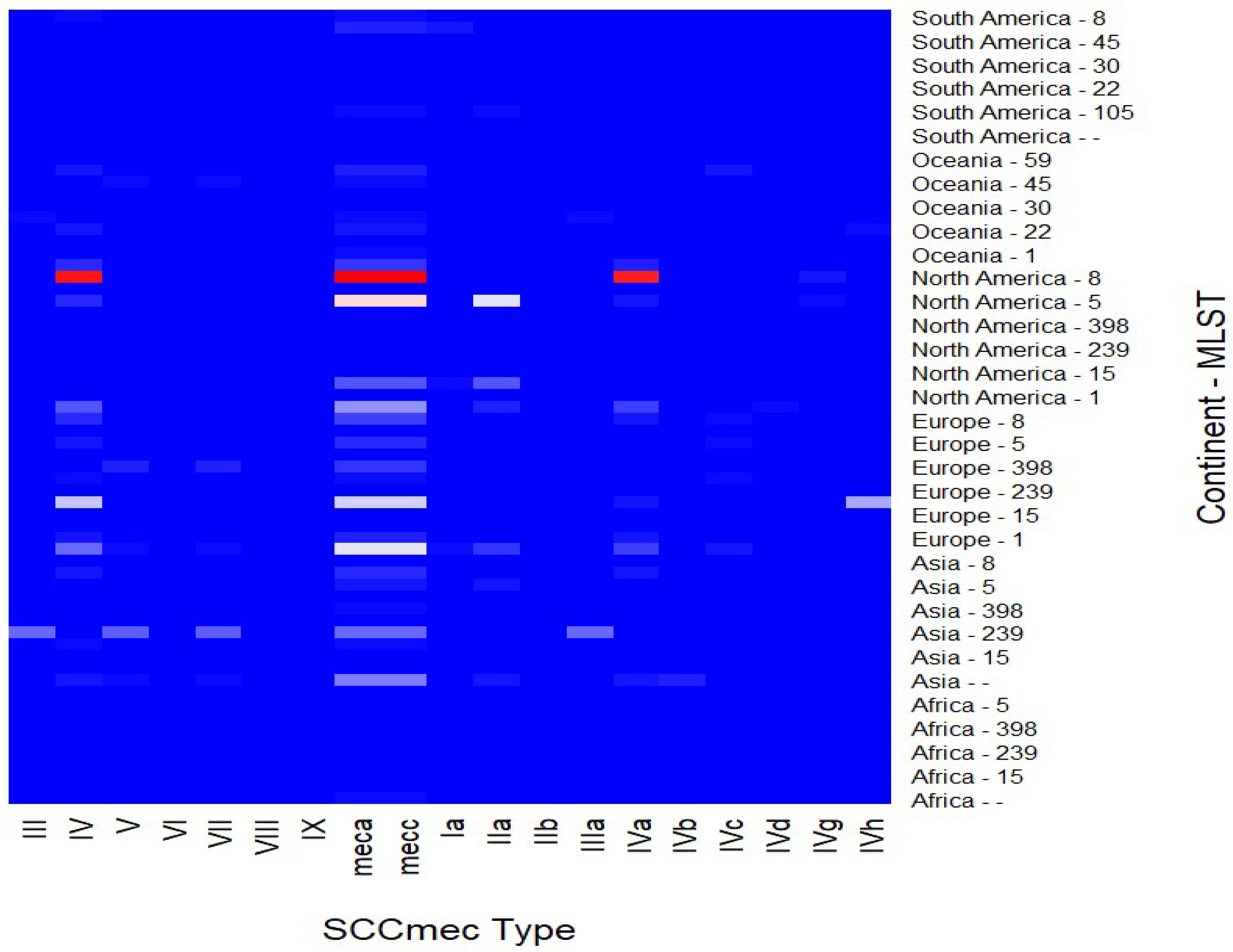
2.7. Global Distribution of SCCmec Types Among MLST Profiles and Continents
3. Discussion
3.1. Distribution of Sequences in the World
3.2. Association of Genes Linked to Biofilm Production and Resistance Genes
3.3. Correlation Between Antimicrobial Resistance Genes and Biofilm-Related Genes
3.4. Acquisition of Antimicrobial Resistance in Clinical Strains
3.5. Acquisition of Antimicrobial Resistance in Environmental Strains
3.6. Prevalent Sequence Types (STs) in Human Clinical and Environmental Samples
3.7. Associations Between SCCmec Types and MLST Profiles
3.8. Perspectives
3.9. Study Limitations
4. Materials and Methods
4.1. Genome Acquisition
4.2. Screening of Metadata Belonging to Genomic Sequences
4.3. Characterization of the Presence and Absence of Genes
4.4. Typing the Genomic Sequences
4.5. Statistical Analysis
- Calculation of gene frequencies.
- Evaluation of genetic correlations.
- Identification of genes with statistically significant associations.
- Analysis of the global distribution of MLST and SCCmec types.
4.6. Samples
Clinical and Environmental
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanssen, A.-M.; Sollid, J.U.E. SCCmecin staphylococci: Genes on the move. FEMS Immunol. Med. Microbiol. 2006, 46, 8–20. [Google Scholar] [CrossRef]
- Lade, H.; Kim, J.-S. Molecular Determinants of β-Lactam Resistance in Methicillin-Resistant Staphylococcus aureus (MRSA): An Updated Review. Antibiotics 2023, 12, 1362. [Google Scholar] [CrossRef] [PubMed]
- Wisplinghoff, H.; Rosato, A.E.; Enright, M.C.; Noto, M.; Craig, W.; Archer, G.L. Related Clones Containing SCC mec Type IV Predominate among Clinically Significant Staphylococcus epidermidis Isolates. Antimicrob. Agents Chemother. 2003, 47, 3574–3579. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.X.; Ito, T.; Tiensasitorn, C.; Jamklang, M.; Chongtrakool, P.; Boyle-Vavra, S.; Daum, R.S.; Hiramatsu, K. Novel Type of Staphylococcal Cassette Chromosome mec Identified in Community-Acquired Methicillin-Resistant Staphylococcus aureus Strains. Antimicrob. Agents Chemother. 2002, 46, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Shore, A.; Rossney, A.S.; Keane, C.T.; Enright, M.C.; Coleman, D.C. Seven Novel Variants of the Staphylococcal Chromosomal Cassette mec in Methicillin-Resistant Staphylococcus aureus Isolates from Ireland. Antimicrob. Agents Chemother. 2005, 49, 2070–2083. [Google Scholar] [CrossRef]
- Boundy, S.; Safo, M.K.; Wang, L.; Musayev, F.N.; O’Farrell, H.C.; Rife, J.P.; Archer, G.L. Characterization of the Staphylococcus aureus rRNA Methyltransferase Encoded by orfX, the Gene Containing the Staphylococcal Chromosome Cassette mec (SCCmec) Insertion Site. J. Biol. Chem. 2013, 288, 132–140. [Google Scholar] [CrossRef]
- Ito, T.; Katayama, Y.; Asada, K.; Ito, T.; Katayama, Y.; Asada, K.; Mori, N.; Tsutsumimoto, K.; Tiensasitorn, C.; Hiramatsu, K. Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1323–1336. [Google Scholar] [CrossRef]
- Maree, M.; Thi Nguyen, L.T.; Ohniwa, R.L.; Higashide, M.; Msadek, T.; Morikawa, K. Natural transformation allows transfer of SCCmec-mediated methicillin resistance in Staphylococcus aureus biofilms. Nat. Commun. 2022, 13, 2477. [Google Scholar] [CrossRef]
- Reischl, U.; Frick, J.; Hoermansdorfer, S.; Melzl, H.; Bollwein, M.; Linde, H.J.; Becker, K.; Köck, R.; Tuschak, C.; Busch, U.; et al. Single-nucleotide polymorphism in the SCCmec-orfX junction distinguishes between livestock-associated MRSA CC398 and human epidemic MRSA strains. Eurosurveillance 2009, 14, 19436. [Google Scholar] [CrossRef]
- Harrison, E.M.; Paterson, G.K.; Holden, M.T.G.; Ba, X.; Rolo, J.; Morgan, F.J.E.; Pichon, B.; Kearns, A.; Zadoks, R.N.; Peacock, S.J.; et al. A novel hybrid SCCmec-mecC region in Staphylococcus sciuri. J. Antimicrob. Chemother. 2014, 69, 911–918. [Google Scholar] [CrossRef]
- Bartels, M.D.; Hansen, L.H.; Boye, K.; Sørensen, S.J.; Westh, H. An Unexpected Location of the Arginine Catabolic Mobile Element (ACME) in a USA300-Related MRSA Strain. PLoS ONE 2011, 6, e16193. [Google Scholar] [CrossRef]
- Chongtrakool, P.; Ito, T.; Ma, X.X.; Kondo, Y.; Trakulsomboon, S.; Tiensasitorn, C.; Jamklang, M.; Chavalit, T.; Song, J.-H.; Hiramatsu, K. Staphylococcal Cassette Chromosome mec (SCC mec) Typing of Methicillin-Resistant Staphylococcus aureus Strains Isolated in 11 Asian Countries: A Proposal for a New Nomenclature for SCC mec Elements. Antimicrob. Agents Chemother. 2006, 50, 1001–1012. [Google Scholar] [CrossRef]
- Gordon, R.J.; Lowy, F.D. Pathogenesis of Methicillin-ResistantStaphylococcus aureusInfection. Clin. Infect. Dis. 2008, 46, S350–S359. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, J.A. Genomic variation and evolution of Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Parisi, A.; Caruso, M.; Normanno, G.; Latorre, L.; Sottili, R.; Miccolupo, A.; Fraccalvieri, R.; Santagada, G. Prevalence, antimicrobial susceptibility and molecular typing of Methicillin-Resistant Staphylococcus aureus (MRSA) in bulk tank milk from southern Italy. Food Microbiol. 2016, 58, 36–42. [Google Scholar] [CrossRef]
- Gutiérrez, D.; Delgado, S.; Vázquez-Sánchez, D.; Martínez, B.; Cabo, M.L.; Rodríguez, A.; Herrera, J.J.; García, P. Incidence of Staphylococcus aureus and analysis of associated bacterial communities on food industry surfaces. Appl. Environ. Microbiol. 2012, 78, 8547–8554. [Google Scholar]
- Vázquez-Sánchez, D.; A Galvão, J.; Oetterer, M. Contamination sources, biofilm-forming ability and biocide resistance of Staphylococcus aureus in tilapia-processing facilities. Food Sci. Technol. Int. 2018, 24, 209–222. [Google Scholar] [CrossRef]
- Liu, P.Y.F.; Gur, D.; Hall, L.M.C.; Livermore, D.M. Survey of the prevalence of βlactamases amongst 1000 Gram-negative bacilli isolated consecutively at the Royal London Hospital. J. Antimicrob. Chemother. 1992, 30, 429–447. [Google Scholar] [CrossRef]
- Di Ciccio, P.; Vergara, A.; Festino, A.; Paludi, D.; Zanardi, E.; Ghidini, S.; Ianieri, A. Biofilm formation by Staphylococcus aureus on food contact surfaces: Relationship with temperature and cell surface hydrophobicity. Food Control 2015, 50, 930–936. [Google Scholar] [CrossRef]
- Abbasi, K.; Tajbakhsh, E.; Momtaz, H. Antimicrobial Resistance and Biofilm Encoding Genes Amongst the Staphylococcus aureus Bacteria Isolated From Meat and Meat Products. NIDOC-ASRT 2021, 52, 55–62. [Google Scholar] [CrossRef]
- Osman, K.; Alvarez-Ordóñez, A.; Ruiz, L.; Badr, J.; ElHofy, F.; Al-Maary, K.S.; Moussa, I.M.I.; Hessain, A.M.; Orabi, A.; Saad, A.; et al. Antimicrobial resistance and virulence characterization of Staphylococcus aureus and coagulase-negative staphylococci from imported beef meat. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 35. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcal Biofilms. Microbiolspec 2018, 6, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Patti, J.M.; Bremell, T.; Krajewska-Pietrasik, D.; Abdelnour, A.; Tarkowski , A.; Rydén, C.; Höök, M. The Staphylococcus aureus collagen adhesin is a virulence determinant in experimental septic arthritis. Infect. Immun. 1994, 62, 152–160. [Google Scholar] [CrossRef]
- Lade, H.; Park, J.H.; Chung, S.H.; Kim, I.H.; Kim, J.-M.; Joo, H.-S.; Kim, J.-S. Biofilm Formation by Staphylococcus aureus Clinical Isolates is Differentially Affected by Glucose and Sodium Chloride Supplemented Culture Media. J. Clin. Med. 2019, 8, 1853. [Google Scholar] [CrossRef]
- Oniciuc, E.-A.; Cerca, N.; Nicolau, A.I. Compositional Analysis of Biofilms Formed by Staphylococcus aureus Isolated from Food Sources. Front. Microbiol. 2016, 7, 390. [Google Scholar] [CrossRef]
- O’Gara, J.P. icaand beyond: Biofilm mechanisms and regulation inStaphylococcus epidermidisandStaphylococcus aureus. FEMS Microbiol. Lett. 2007, 270, 179–188. [Google Scholar] [CrossRef]
- Maira-Litrán, T.; Kropec, A.; Abeygunawardana, C.; Joyce, J.; Mark, G.; Goldmann, D.A.; Pier, G.B. Immunochemical Properties of the Staphylococcal Poly- N -Acetylglucosamine Surface Polysaccharide. Infect. Immun. 2002, 70, 4433–4440. [Google Scholar] [CrossRef]
- Liu, J.; Yang, L.; Hou, Y.; Soteyome, T.; Zeng, B.; Su, J.; Li, L.; Li, B.; Chen, D.; Li, Y.; et al. Transcriptomics Study on Staphylococcus aureus Biofilm Under Low Concentration of Ampicillin. Front. Microbiol. 2018, 9, 2413. [Google Scholar] [CrossRef] [PubMed]
- Herman-Bausier, P.; El-Kirat-Chatel, S.; Foster, T.J.; Geoghegan, J.A.; Dufrêne, Y.F. Staphylococcus aureus Fibronectin-Binding Protein A Mediates Cell-Cell Adhesion through Low-Affinity Homophilic Bonds. mBio 2015, 6, e00413-15. [Google Scholar] [CrossRef]
- O’Brien, L.M.; Walsh, E.J.; Massey, R.C.; Peacock, S.J.; Foster, T.J. Staphylococcus aureusclumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: Implications for nasal colonization. Cell. Microbiol. 2002, 4, 759–770. [Google Scholar] [CrossRef]
- Cucarella, C.; Solano, C.; Valle, J.; Amorena, B.; Lasa, Í.; Penadés, J.R. Bap, a Staphylococcus aureus Surface Protein Involved in Biofilm Formation. J. Bacteriol. 2001, 183, 2888–2896. [Google Scholar] [CrossRef] [PubMed]
- Barbu, E.M.; Mackenzie, C.; Foster, T.J.; Höök, M. SdrC induces staphylococcal biofilm formation through a homophilic interaction. Mol. Microbiol. 2014, 94, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Campoccia, D.; Montanaro, L.; Ravaioli, S.; Cangini, I.; Speziale, P.; Arciola, C.R. Description of a New Group of Variants of the Staphylococcus Aureus Elastin-Binding Protein that Lacks an Entire DNA Segment of 180 bp. Int. J. Artif. Organs 2009, 32, 621–629. [Google Scholar] [CrossRef]
- Kranjec, C.; Angeles, D.M.; Mårli, M.T.; Fernández, L.; García, P.; Kjos, M.; Diep, D.B. Staphylococcal Biofilms: Challenges and Novel Therapeutic Perspectives. Antibiotics 2021, 10, 131. [Google Scholar] [CrossRef]
- Yonemoto, K.; Chiba, A.; Sugimoto, S.; Sato, C.; Saito, M.; Kinjo, Y.; Marumo, K.; Mizunoe, Y. Redundant and Distinct Roles of Secreted Protein Eap and Cell Wall-Anchored Protein SasG in Biofilm Formation and Pathogenicity of Staphylococcus aureus. Infect. Immun. 2019, 87, e00894-18. [Google Scholar] [CrossRef]
- Loughran, A.J.; Atwood, D.N.; Anthony, A.C.; Harik, N.S.; Spencer, H.J.; Beenken, K.E.; Smeltzer, M.S. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. Microbiologyopen 2014, 3, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Cuellar, E.; Tsuchiya, K.; Valle-Ríos, R.; Medina-Contreras, O. Differences in Biofilm Formation by Methicillin-Resistant and Methicillin-Susceptible Staphylococcus aureus Strains. Diseases 2023, 11, 160. [Google Scholar] [CrossRef]
- Rodríguez-Lázaro, D.; Alonso-Calleja, C.; Oniciuc, E.A.; Capita, R.; Gallego, D.; González-Machado, C.; Wagner, M.; Barbu, V.; Eiros-Bouza, J.M.; Nicolau, A.I.; et al. Characterization of Biofilms Formed by Foodborne Methicillin-Resistant Staphylococcus aureus. Front. Microbiol. 2018, 9, 3004. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Wang, Y.; Sun, Y.; Ma, L.; Zeng, Q.; Jiang, X.; Li, A.; Zeng, Z.; Zhang, T. Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome 2018, 6, 34. [Google Scholar] [CrossRef]
- Novick, R.P.; Schlievert, P.M.; Ruzin, A. Pathogenicity and resistance islands of staphylococci. Microbes Infect. 2001, 3, 585–594. [Google Scholar] [CrossRef]
- Queck, S.Y.; Khan, B.A.; Wang, R.; Bach, T.-H.L.; Kretschmer, D.; Chen, L.; Kreiswirth, B.N.; Peschel, A.; DeLeo, F.R.; Otto, M. Mobile Genetic Element-Encoded Cytolysin Connects Virulence to Methicillin Resistance in MRSA. PLoS Pathog. 2009, 5, e1000533. [Google Scholar] [CrossRef]
- Firth, N.; Jensen, S.O.; Kwong, S.M.; Skurray, R.A.; Ramsay, J.P. Staphylococcal Plasmids, Transposable and Integrative Elements. Microbiol. Spectr. 2018, 6, 10–1128. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Zhang, F.; Wu, S.; Huang, J.; Yang, R.; Zhang, J.; Lei, T.; Dai, J.; Ding, Y.; Xue, L.; Wang, J.; et al. Presence and Characterization of a Novel cfr-Carrying Tn558 Transposon Derivative in Staphylococcus delphini Isolated From Retail Food. Front. Microbiol. 2021, 11, 598990. [Google Scholar] [CrossRef]
- Mlynarczyk-Bonikowska, B.; Kowalewski, C.; Krolak-Ulinska, A.; Marusza, W. Molecular Mechanisms of Drug Resistance in Staphylococcus aureus. Int. J. Mol. Sci. 2022, 23, 8088. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Ram, G. The Floating (Pathogenicity) Island: A Genomic Dessert. Trends Genet. 2016, 32, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Savage, V.J.; Chopra, I.; O’Neill, A.J. Staphylococcus aureus Biofilms Promote Horizontal Transfer of Antibiotic Resistance. Antimicrob. Agents Chemother. 2013, 57, 1968–1970. [Google Scholar] [CrossRef]
- Iurescia, M.; Diaconu, E.L.; Alba, P.; Feltrin, F.; Buccella, C.; Onorati, R.; Giacomi, A.; Caprioli, A.; Franco, A.; Battisti, A.; et al. Genomics Insight into cfr-Mediated Linezolid-Resistant LA-MRSA in Italian Pig Holdings. Antibiotics 2023, 12, 530. [Google Scholar] [CrossRef]
- Shore, A.C.; Brennan, O.M.; Ehricht, R.; Monecke, S.; Schwarz, S.; Slickers, P.; Coleman, D.C. Identification and Characterization of the Multidrug Resistance Gene cfr in a Panton-Valentine Leukocidin-Positive Sequence Type 8 Methicillin-Resistant Staphylococcus aureus IVa (USA300) Isolate. Antimicrob. Agents Chemother. 2010, 54, 4978–4984. [Google Scholar] [CrossRef]
- Kehrenberg, C.; Schwarz, S. fexA, a Novel Staphylococcus lentus Gene Encoding Resistance to Florfenicol and Chloramphenicol. Antimicrob. Agents Chemother. 2004, 48, 615–618. [Google Scholar] [CrossRef]
- Wolska-Gębarzewska, M.; Międzobrodzki, J.; Kosecka-Strojek, M. Current types of staphylococcal cassette chromosome mec (SCC mec) in clinically relevant coagulase-negative staphylococcal (CoNS) species. Crit. Rev. Microbiol. 2023, 50, 1020–1036. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-M.; Huang, M.; Chen, H.-F.; Ke, S.-C.; Li, C.-R.; Wang, J.-H.; Wu, L.-T. Fusidic acid resistance among clinical isolates of methicillin-resistant Staphylococcus aureus in a Taiwanese hospital. BMC Microbiol. 2011, 11, 98. [Google Scholar] [CrossRef]
- Gupta, S.K.; Pfeltz, R.F.; Wilkinson, B.J.; Gustafson, J.E. Transcriptomic and Metabolomic Analysis of a Fusidic Acid-Selected fusA Mutant of Staphylococcus aureus. Antibiotics 2022, 11, 1051. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, X.; Wang, B.; Xu, Y.; Rao, L.; Wan, B.; Guo, Y.; Wu, X.; Yu, J.; Chen, L.; et al. The Prevalence and Determinants of Fusidic Acid Resistance Among Methicillin-Resistant Staphylococcus aureus Clinical Isolates in China. Front. Med. 2021, 8, 761894. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.A.; Carter, G.P.; Howden, B.P. Current and Emerging Topical Antibacterials and Antiseptics: Agents, Action, and Resistance Patterns. Clin. Microbiol. Rev. 2017, 30, 827–860. [Google Scholar] [CrossRef] [PubMed]
- O’brien, F.G.; Price, C.; Grubb, W.B.; Gustafson, J.E. Genetic characterization of the fusidic acid and cadmium resistance determinants of Staphylococcus aureus plasmid pUB101. J. Antimicrob. Chemother. 2002, 50, 313–321. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-Time Whole-Genome Sequencing for Routine Typing, Surveillance, and Outbreak Detection of Verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.G.; Joensen, K.G.; Østerlund, M.T.; Kiil, K.; Nielsen, E.M. Prediction of antimicrobial resistance in clinical Campylobacter jejuni isolates from whole-genome sequencing data. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 40, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, R.S.; Munk, P.; Njage, P.; Van Bunnik, B.; McNally, L.; Lukjancenko, O.; Röder, T.; Nieuwenhuijse, D.; Pedersen, S.K.; Kjeldgaard, J.; et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 2019, 10, 1124. [Google Scholar] [CrossRef]
- Kleinheinz, K.A.; Joensen, K.G.; Larsen, M.V. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 2014, 4, e27943. [Google Scholar] [CrossRef]
- Egli, A.; Schrenzel, J.; Greub, G. Digital microbiology. Clin. Microbiol. Infect. 2020, 26, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Petit, R.A.; Read, T.D. Staphylococcus aureus viewed from the perspective of 40,000+ genomes. PeerJ 2018, 6, e5261. [Google Scholar] [CrossRef]
- Lindsay, J.A. Staphylococci: Evolving Genomes. Microbiol. Spectr. 2019, 7, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rivas, J.D.L.; Bonavides-Martínez, C.; Campos-Laborie, F.J. Bioinformatics in Latin America and SoIBio impact, a tale of spin-off and expansion around genomes and protein structures. Brief. Bioinform. 2019, 20, 390–397. [Google Scholar] [CrossRef]
- Van Boeckel, T.P.; Pires, J.; Silvester, R.; Zhao, C.; Song, J.; Criscuolo, N.G.; Gilbert, M.; Bonhoeffer, S.; Laxminarayan, R. Global trends in antimicrobial resistance in animals in low- and middle-income countries. Science 2019, 365, eaaw1944. [Google Scholar] [CrossRef]
- Missiakas, D.M.; Schneewind, O. Growth and Laboratory Maintenance of Staphylococcus aureus. Curr. Protoc. Microbiol. 2013, 28, 9C-1. [Google Scholar] [CrossRef]
- Strauß, L.; Stegger, M.; Akpaka, P.E.; Alabi, A.; Breurec, S.; Coombs, G.; Egyir, B.; Larsen, A.R.; Laurent, F.; Monecke, S.; et al. Origin, evolution, and global transmission of community-acquired Staphylococcus aureus ST8. Proc. Natl. Acad. Sci. USA 2017, 114, E10596–E10604. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Gray, G.C. The Mandate for a Global “One Health” Approach to Antimicrobial Resistance Surveillance. Am. J. Trop. Med. Hyg. 2019, 100, 227–228. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, T.P.; Brower, C.; Gilbert, M.; Grenfell, B.T.; Levin, S.A.; Robinson, T.P.; Teillant, A.; Laxminarayan, R. Global trends in antimicrobial use in food animals. Proc. Natl. Acad. Sci. USA 2015, 112, 5649–5654. [Google Scholar] [CrossRef]
- Conceição, S.; Queiroga, M.C.; Laranjo, M. Antimicrobial Resistance in Bacteria from Meat and Meat Products: A One Health Perspective. Microorganisms 2023, 11, 2581. [Google Scholar] [CrossRef]
- Silva, A.C.; Ferrari, R.G.; Panzenhagen, P.; Conte, C.A. Staphylococcus aureus biofilm: The role in disseminating antimicrobial resistance over the meat chain. Microbiology 2022, 168, 001245. [Google Scholar]
- Urban-Chmiel, R.; Marek, A.; Stępień-Pyśniak, D.; Wieczorek, K.; Dec, M.; Nowaczek, A.; Osek, J. Antibiotic Resistance in Bacteria—A Review. Antibiotics 2022, 11, 1079. [Google Scholar] [CrossRef]
- Aslam, B.; Khurshid, M.; Arshad, M.I.; Muzammil, S.; Rasool, M.; Yasmeen, N.; Shah, T.; Chaudhry, T.H.; Rasool, M.H.; Shahid, A.; et al. Antibiotic Resistance: One Health One World Outlook. Front. Cell. Infect. Microbiol. 2021, 11, 771510. [Google Scholar] [CrossRef]
- Algammal, A.M.; Hetta, H.F.; Elkelish, A.; Alkhalifah, D.H.H.; Hozzein, W.N.; Batiha, G.E.-S.; El Nahhas, N.; A Mabrok, M. Methicillin-Resistant Staphylococcus aureus (MRSA): One Health Perspective Approach to the Bacterium Epidemiology, Virulence Factors, Antibiotic-Resistance, and Zoonotic Impact. Infect. Drug Resist. 2020, 13, 3255–3265. [Google Scholar] [CrossRef]
- Reyes, J.; Rincón, S.; Díaz, L.; Panesso, D.; Contreras, G.A.; Zurita, J.; Carrillo, C.; Rizzi, A.; Guzmán, M.; Adachi, J.; et al. Dissemination of Methicillin-ResistantStaphylococcus aureusUSA300 Sequence Type 8 Lineage in Latin America. Clin. Infect. Dis. 2009, 49, 1861–1867. [Google Scholar] [CrossRef]
- Barcudi, D.; Blasko, E.; Gonzalez, M.J.; Gagetti, P.; Lamberghini, R.; Garnero, A.; Sarkis, C.; Faccone, D.; Lucero, C.; Tosoroni, D.; et al. Different evolution of S. aureus methicillin-resistant and methicillin-susceptible infections, Argentina. Heliyon 2024, 10, e22610. [Google Scholar] [CrossRef]
- Saba, C.K.S.; Amenyona, J.K.; Kpordze, S.W. Prevalence and pattern of antibiotic resistance of Staphylococcus aureus isolated from door handles and other points of contact in public hospitals in Ghana. Antimicrob. Resist. Infect. Control. 2017, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Nsour, E.H.A.; Al-Hadithi, H.T.; Al-Groom, R.M.; Abushattal, S.; Naser, A.Y.; Al Nsour, A.H.; Sallam, R.A.; Kollab, L.M.; Alswalha, L.; Khan, M.S.A. Increased incidence of methicillin resistant Staphylococcus aureus and methicillin resistant Staphylococcus epidermidis in the skin and nasal carriage among healthcare workers and inanimate hospital surfaces after the COVID-19 pandemic. Iran. J. Microbiol. 2024, 16, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Kinnevey, P.; Kearney, A.; Shore, A.; Earls, M.; Brennan, G.; Poovelikunnel, T.; Humphreys, H.; Coleman, D. Meticillin-susceptible Staphylococcus aureus transmission among healthcare workers, patients and the environment in a large acute hospital under non-outbreak conditions investigated using whole-genome sequencing. J. Hosp. Infect. 2022, 127, 15–25. [Google Scholar] [CrossRef]
- Price, J.R.; Cole, K.; Bexley, A.; Kostiou, V.; Eyre, D.W.; Golubchik, T.; Wilson, D.J.; Crook, D.W.; Walker, A.S.; E A Peto, T.; et al. Transmission of Staphylococcus aureus between health-care workers, the environment, and patients in an intensive care unit: A longitudinal cohort study based on whole-genome sequencing. Lancet Infect. Dis. 2017, 17, 207–214. [Google Scholar] [CrossRef]
- Hao, H.; Cheng, G.; Iqbal, Z.; Ai, X.; Hussain, H.I.; Huang, L.; Dai, M.; Wang, Y.; Liu, Z.; Yuan, Z.-H. Benefits and risks of antimicrobial use in food-producing animals. Front. Microbiol. 2014, 5, 288. [Google Scholar] [CrossRef]
- Kimera, Z.I.; Mshana, S.E.; Rweyemamu, M.M.; Mboera, L.E.G.; Matee, M.I.N. Antimicrobial use and resistance in food-producing animals and the environment: An African perspective. Antimicrob. Resist. Infect. Control 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Chen, B.-H.; Kuca, K.; Nepovimova, E.; Kaushal, A.; Nagraik, R.; Bhatia, S.K.; Dhanjal, D.S.; Kumar, V.; Kumar, A.; et al. Understanding of Colistin Usage in Food Animals and Available Detection Techniques: A Review. Animals 2020, 10, 1892. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.; Wu, S.; Zhang, F.; Huang, J.; Wu, H.; Zhang, J.; Li, Y.; Ding, Y.; Zhang, J.; Chen, M.; et al. The Genomic Context for the Evolution and Transmission of Community-Associated Staphylococcus aureus ST59 Through the Food Chain. Front. Microbiol. 2020, 11, 422. [Google Scholar] [CrossRef]
- Becker, K.; Ballhausen, B.; Kahl, B.C.; Köck, R. The clinical impact of livestock-associated methicillin-resistant Staphylococcus aureus of the clonal complex 398 for humans. Vet. Microbiol. 2017, 200, 33–38. [Google Scholar] [CrossRef]
- Cazer, C.L.; Eldermire, E.R.; Lhermie, G.; Murray, S.A.; Scott, H.M.; Gröhn, Y.T. The effect of tylosin on antimicrobial resistance in beef cattle enteric bacteria: A systematic review and meta-analysis. Prev. Vet. Med. 2020, 176, 104934. [Google Scholar] [CrossRef]
- Kruse, A.B.; Kristensen, C.S.; Lavlund, U.; Stege, H. Antimicrobial prescription data in Danish national database validated against treatment records in organic pig farms and analysed for associations with lesions found at slaughter. BMC Vet. Res. 2019, 15, 218. [Google Scholar] [CrossRef]
- Lowder, B.V.; Guinane, C.M.; Ben Zakour, N.L.; Weinert, L.A.; Conway-Morris, A.; Cartwright, R.A.; Simpson, A.J.; Rambaut, A.; Nübel, U.; Fitzgerald, J.R. Recent human-to-poultry host jump, adaptation, and pandemic spread of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2009, 106, 19545–19550. [Google Scholar] [CrossRef]
- Moufti, M.F.E.; Baddour, M.; Harfoush, R.A.H.; Owais, H.M.A. Characterization of Some Genotypic and Phenotypic Traits of Biofilm Producing Clinical Isolates of Methicillin Resistant Staphylococcus Epidermidis. Am. J. Infect. Dis. Microbiol. 2019, 17, 100444. [Google Scholar]
- Skaar, E.P.; Schneewind, O. Iron-regulated surface determinants (Isd) of Staphylococcus aureus: Stealing iron from heme. Microbes Infect. 2004, 6, 390–397. [Google Scholar] [CrossRef]
- Alkuraythi, D.M.; Alkhulaifi, M.M.; Binjomah, A.Z.; Alarwi, M.; Mujallad, M.I.; Alharbi, S.A.; Alshomrani, M.; Gojobori, T.; Alajel, S.M. Comparative genomic analysis of antibiotic resistance and virulence genes in Staphylococcus aureus isolates from patients and retail meat. Front. Cell. Infect. Microbiol. 2024, 13, 1339339. [Google Scholar] [CrossRef]
- Grigg, J.C.; Ukpabi, G.; Gaudin, C.F.; Murphy, M.E. Structural biology of heme binding in the Staphylococcus aureus Isd system. J. Inorg. Biochem. 2010, 104, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Speziale, P.; Pietrocola, G.; Foster, T.J.; Geoghegan, J.A. Protein-based biofilm matrices in Staphylococci. Front. Cell. Infect. Microbiol. 2014, 4, 171. [Google Scholar] [CrossRef]
- Corrigan, R.M.; Miajlovic, H.; Foster, T.J. Surface proteins that promote adherence of Staphylococcus aureus to human desquamated nasal epithelial cells. BMC Microbiol. 2009, 9, 22. [Google Scholar]
- Yu, F.; Cienfuegos-Gallet, A.V.; Cunningham, M.H.; Jin, Y.; Wang, B.; Kreiswirth, B.N.; Chen, L. Molecular Evolution and Adaptation of Livestock-Associated Methicillin-Resistant Staphylococcus aureus (LA-MRSA) Sequence Type 9. Msystems 2021, 6, e0049221. [Google Scholar] [CrossRef]
- Ibrahim, E.S.; Arafa, A.A.; Dorgam, S.M.; Eid, R.H.; Atta, N.S.; El-Dabae, W.H.; Gaber, E.S. Molecular characterization of genes responsible for biofilm formation in Staphylococcus aureus isolated from mastitic cows. Vet. World 2022, 15, 205–212. [Google Scholar] [CrossRef]
- Moretto, V.T.; Bartley, P.S.; Ferreira, V.M.; Santos, C.S.; Silva, L.K.; Ponce-Terashima, R.A.; Blanton, R.E.; Reis, M.G.; Barbosa, L.M. Microbial source tracking and antimicrobial resistance in one river system of a rural community in Bahia, Brazil. Braz. J. Biol. 2022, 82, e231838. [Google Scholar] [CrossRef]
- Carrera-Salinas, A.; González-Díaz, A.; Vázquez-Sánchez, D.A.; Camoez, M.; Niubó, J.; Càmara, J.; Ardanuy, C.; Martí, S.; Domínguez, M.Á.; Garcia, M.; et al. Staphylococcus aureus surface protein G (sasG) allelic variants: Correlation between biofilm formation and their prevalence in methicillin-resistant S. aureus (MRSA) clones. Res. Microbiol. 2022, 173, 103921. [Google Scholar] [CrossRef]
- Missineo, A.; Di Poto, A.; Geoghegan, J.A.; Rindi, S.; Heilbronner, S.; Gianotti, V.; Arciola, C.R.; Foster, T.J.; Speziale, P.; Pietrocola, G. IsdC from Staphylococcus lugdunensis Induces Biofilm Formation under Low-Iron Growth Conditions. Infect. Immun. 2014, 82, 2448–2459. [Google Scholar] [CrossRef]
- Andrade, M.M.; Luiz, W.B.; Souza, R.d.S.O.; Amorim, J.H. The History of Methicillin-Resistant Staphylococcus aureus in Brazil. Can. J. Infect. Dis. Med. Microbiol. 2020, 2020, 1721936. [Google Scholar] [CrossRef]
- Banerjee, P.; Eulenstein, O.; Friedberg, I. Discovering genomic islands in unannotated bacterial genomes using sequence embedding. Bioinform. Adv. 2024, 4, vbae089. [Google Scholar] [CrossRef]
- Monecke, S.; Roberts, M.C.; Braun, S.D.; Diezel, C.; Müller, E.; Reinicke, M.; Linde, J.; Joshi, P.R.; Paudel, S.; Acharya, M.; et al. Sequence Analysis of Novel Staphylococcus aureus Lineages from Wild and Captive Macaques. Int. J. Mol. Sci. 2022, 23, 11225. [Google Scholar] [CrossRef]
- Mendes, R.E.; Deshpande, L.M.; Castanheira, M.; DiPersio, J.; Saubolle, M.A.; Jones, R.N. First Report of cfr -Mediated Resistance to Linezolid in Human Staphylococcal Clinical Isolates Recovered in the United States. Antimicrob. Agents Chemother. 2008, 52, 2244–2246. [Google Scholar] [CrossRef]
- Vogt, D.; Overesch, G.; Endimiani, A.; Collaud, A.; Thomann, A.; Perreten, V. Occurrence and Genetic Characteristics of Third-Generation Cephalosporin-Resistant Escherichia coli in Swiss Retail Meat. Microb. Drug Resist. 2014, 20, 485–494. [Google Scholar] [CrossRef]
- Werth, B.J.; Steed, M.E.; Ireland, C.E.; Tran, T.T.; Nonejuie, P.; Murray, B.E.; Rose, W.E.; Sakoulas, G.; Pogliano, J.; Arias, C.A.; et al. Defining Daptomycin Resistance Prevention Exposures in Vancomycin-Resistant Enterococcus faecium and E. faecalis. Antimicrob. Agents Chemother. 2014, 58, 5253–5261. [Google Scholar] [CrossRef] [PubMed]
- Monecke, S.; Gavier-Widén, D.; Hotzel, H.; Peters, M.; Guenther, S.; Lazaris, A.; Loncaric, I.; Müller, E.; Reissig, A.; Ruppelt-Lorz, A.; et al. Diversity of Staphylococcus aureus Isolates in European Wildlife. PLoS ONE 2016, 11, e0168433. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y. Current Status of Staphylococcal Cassette Chromosome mec (SCCmec). Antibiotics 2022, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.A.; Baig, F.K.; Mehboob, R. Nosocomial infections: Epidemiology, prevention, control and surveillance. Asian Pac. J. Trop. Biomed. 2017, 7, 478–482. [Google Scholar] [CrossRef]
- Casanova, N.G.; Ruiz, M.S.; Bellido, J.L.M. Mechanisms of resistance to daptomycin in Staphylococcus aureus. Rev. Esp. Quimioter 2017, 30, 391–396. [Google Scholar]
- Prater, A.G.; Mehta, H.H.; Kosgei, A.J.; Miller, W.R.; Tran, T.T.; Arias, C.A.; Shamoo, Y. Environment Shapes the Accessible Daptomycin Resistance Mechanisms in Enterococcus faecium. Antimicrob. Agents Chemother. 2019, 63, e00790-19. [Google Scholar] [CrossRef]
- Adator, E.H.; Narvaez-Bravo, C.; Zaheer, R.; Cook, S.R.; Tymensen, L.; Hannon, S.J.; Booker, C.W.; Church, D.; Read, R.R.; McAllister, T.A. A One Health Comparative Assessment of Antimicrobial Resistance in Generic and Extended-Spectrum Cephalosporin-Resistant Escherichia coli from Beef Production, Sewage and Clinical Settings. Microorganisms 2020, 8, 885. [Google Scholar] [CrossRef]
- Deurenberg, R.; Vink, C.; Kalenic, S.; Friedrich, A.; Bruggeman, C.; Stobberingh, E. The molecular evolution of methicillin-resistant Staphylococcus aureus. Clin. Microbiol. Infect. 2007, 13, 222–235. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.T.; Sharma, S.; Sivakumar, N.; Jayakumar, K. Genomic islands and the evolution of livestock-associated Staphylococcus aureus genomes. Biosci. Rep. 2020, 40, BSR20202287. [Google Scholar] [PubMed]
- Silbergeld, E.K.; Graham, J.; Price, L.B. Industrial food animal production, antimicrobial resistance, and human health. Annu. Rev. Public Health 2008, 29, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef]
- Buelow, E.; Ploy, M.-C.; Dagot, C. Role of pollution on the selection of antibiotic resistance and bacterial pathogens in the environment. Curr. Opin. Microbiol. 2021, 64, 117–124. [Google Scholar] [CrossRef]
- Miragaia, M. Factors Contributing to the Evolution of mecA-Mediated β-lactam Resistance in Staphylococci: Update and New Insights From Whole Genome Sequencing (WGS). Front. Microbiol. 2018, 9, 2723. [Google Scholar] [CrossRef]
- Nübel, U.; Roumagnac, P.; Feldkamp, M.; Song, J.-H.; Ko, K.S.; Huang, Y.-C.; Coombs, G.; Ip, M.; Westh, H.; Skov, R.; et al. Frequent emergence and limited geographic dispersal of methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2008, 105, 14130–14135. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Koide, S.; Maeyama, Y.; Tamai, K.; Hayashi, W.; Tanaka, H.; Iimura, M.; Suzuki, M.; Nagano, Y.; Arakawa, Y.; et al. Predominance of methicillin-resistant Staphylococcus aureus SCCmec type II-CC5 and SCCmec type IV-CC1/CC8 among companion animal clinical isolates in Japan: Findings from phylogenetic comparison with human clinical isolates. J. Glob. Antimicrob. Resist. 2019, 20, 253–259. [Google Scholar] [CrossRef]
- Martínez, J.R.W.; Planet, P.J.; Spencer-Sandino, M.; Rivas, L.; Díaz, L.; Moustafa, A.M.; Quesille-Villalobos, A.; Riquelme-Neira, R.; Alcalde-Rico, M.; Hanson, B.; et al. Dynamics of the MRSA Population in a Chilean Hospital: A Phylogenomic Analysis (2000–2016). Microbiol. Spectr. 2023, 11, e0535122. [Google Scholar] [CrossRef]
- Li, X.; Zhang, J.; Zhang, Y.; Zhou, J.; Li, X.; Feng, R.; Li, Y. Methicillin-resistant Staphylococcus aureus of the clonal lineage ST5-SCCmecII-t2460 was associated with high mortality in a Wuhan hospital. Braz. J. Microbiol. 2021, 52, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Álvarez, S.; Châtre, P.; François, P.; Abdullahi, I.N.; Simón, C.; Zarazaga, M.; Madec, J.-Y.; Haenni, M.; Torres, C. Unexpected role of pig nostrils in the clonal and plasmidic dissemination of extended-spectrum beta-lactamase-producing Escherichia coli at farm level. Ecotoxicol. Environ. Saf. 2024, 273, 116145. [Google Scholar] [CrossRef] [PubMed]
- Soliman, M.S.; Soliman, N.S.; El-Manakhly, A.R.; ElBanna, S.A.; Aziz, R.K.; El-Kholy, A.A. Genomic Characterization of Methicillin-Resistant Staphylococcus aureus (MRSA) by High-Throughput Sequencing in a Tertiary Care Hospital. Genes 2020, 11, 1219. [Google Scholar] [CrossRef]
- Heaton, C.J.; Gerbig, G.R.; Sensius, L.D.; Patel, V.; Smith, T.C. Staphylococcus aureus Epidemiology in Wildlife: A Systematic Review. Antibiotics 2020, 9, 89. [Google Scholar] [CrossRef]
- Al-Talib, H.; Samsudin, S.; Adnan, A.; Murugaiah, C. Genetic Diversity among Methicillin-Resistant Staphylococcus aureus in Malaysia (2002–2020). Trop. Med. Infect. Dis. 2022, 7, 360. [Google Scholar] [CrossRef] [PubMed]
- Knight, G.M.; Budd, E.L.; Whitney, L.; Thornley, A.; Al-Ghusein, H.; Planche, T.; Lindsay, J.A. Shift in dominant hospital-associated methicillin-resistant Staphylococcus aureus (HA-MRSA) clones over time. J. Antimicrob. Chemother. 2012, 67, 2514–2522. [Google Scholar] [CrossRef]
- Farook, N.A.M.; Argimón, S.; Samat, M.N.A.; Salleh, S.A.; Sulaiman, S.; Tan, T.L.; Periyasamy, P.; Lau, C.L.; Ismail, Z.; Azami, N.A.M.; et al. Diversity and Dissemination of Methicillin-Resistant Staphylococcus aureus (MRSA) Genotypes in Southeast Asia. Trop. Med. Infect. Dis. 2022, 7, 438. [Google Scholar] [CrossRef]
- Monecke, S.; Slickers, P.; Gawlik, D.; Müller, E.; Reissig, A.; Ruppelt-Lorz, A.; Akpaka, P.E.; Bandt, D.; Bes, M.; Boswihi, S.S.; et al. Molecular Typing of ST239-MRSA-III From Diverse Geographic Locations and the Evolution of the SCCmec III Element During Its Intercontinental Spread. Front. Microbiol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Abimanyu, N.; Murugesan, S.; Krishnan, P. Emergence of Methicillin-Resistant Staphylococcus aureus ST239 with High-Level Mupirocin and Inducible Clindamycin Resistance in a Tertiary Care Center in Chennai, South India. J. Clin. Microbiol. 2012, 50, 3412–3413. [Google Scholar] [CrossRef]
- El-Hamid, M.I.A.; Bendary, M.M.; Merwad, A.M.A.; Elsohaby, I.; Ghaith, D.M.; Alshareef, W.A. What is behind phylogenetic analysis of hospital—community and livestock-associated methicillin-resistant Staphylococcus aureus? Transbound. Emerg. Dis. 2019, 66, 1506–1517. [Google Scholar]
- Chen, C.-J.; Unger, C.; Hoffmann, W.; Lindsay, J.A.; Huang, Y.-C.; Götz, F. Characterization and Comparison of 2 Distinct Epidemic Community-Associated Methicillin-Resistant Staphylococcus aureus Clones of ST59 Lineage. PLoS ONE 2013, 8, e63210. [Google Scholar] [CrossRef] [PubMed]
- Kadlec, K.; Schwarz, S. Identification of a Novel Trimethoprim Resistance Gene, dfrK, in a Methicillin-Resistant Staphylococcus aureus ST398 Strain and Its Physical Linkage to the Tetracycline Resistance Gene tet (L). Antimicrob. Agents Chemother. 2009, 53, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Carrel, M.; Shi, Q.; Hasegawa, S.; Clore, G.S.; David, M.Z.; Perencevich, E.N.; Smith, M.; Goto, M. Persistence of potential ST398 MSSA in outpatient settings among US veterans, 2010–2019. ASHE 2023, 3, e177. [Google Scholar] [CrossRef]
- Price, L.B.; Stegger, M.; Hasman, H.; Aziz, M.; Larsen, J.; Andersen, P.S.; Pearson, T.; Waters, A.E.; Foster, J.T.; Schupp, J.; et al. Staphylococcus aureus CC398: Host Adaptation and Emergence of Methicillin Resistance in Livestock. mBio 2012, 3, e00305-11. [Google Scholar] [CrossRef]
- Matuszewska, M.; Murray, G.G.; Ba, X.; Wood, R.; A Holmes, M.; A Weinert, L.; Kingdom, U. Stable antibiotic resistance and rapid human adaptation in livestock-associated MRSA. eLife 2022, 11, e74819. [Google Scholar] [CrossRef]
- Enright, M.C.; Robinson, D.A.; Randle, G.; Feil, E.J.; Grundmann, H.; Spratt, B.G. The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc. Natl. Acad. Sci. USA 2002, 99, 7687–7692. [Google Scholar] [CrossRef] [PubMed]
- Vandenesch, F.; Naimi, T.; Enright, M.C.; Lina, G.; Nimmo, G.R.; Heffernan, H.; Liassine, N.; Bes, M.; Greenland, T.; Reverdy, M.-E.; et al. Community-Acquired Methicillin-ResistantStaphylococcus aureusCarrying Panton-Valentine Leukocidin Genes: Worldwide Emergence. Emerg. Infect. Dis. 2003, 9, 978–984. [Google Scholar] [CrossRef]
- Ho, P.-L.; Cheung, C.; Mak, G.C.; Tse, C.W.; Ng, T.-K.; Cheung, C.H.; Que, T.-L.; Lam, R.; Lai, R.W.; Yung, R.W.; et al. Molecular epidemiology and household transmission of community-associated methicillin-resistant Staphylococcus aureus in Hong Kong. Diagn. Microbiol. Infect. Dis. 2007, 57, 145–151. [Google Scholar] [CrossRef]
- McClure, J.-A.; Lakhundi, S.; Niazy, A.; Dong, G.; Obasuyi, O.; Gordon, P.; Chen, S.; Conly, J.M.; Zhang, K. Staphylococcus aureus ST59: Concurrent but Separate Evolution of North American and East Asian Lineages. Front. Microbiol. 2021, 12, 631845. [Google Scholar] [CrossRef]
- Wang, W.; Baker, M.; Hu, Y.; Xu, J.; Yang, D.; Maciel-Guerra, A.; Xue, N.; Li, H.; Yan, S.; Li, M.; et al. Whole-Genome Sequencing and Machine Learning Analysis of Staphylococcus aureus from Multiple Heterogeneous Sources in China Reveals Common Genetic Traits of Antimicrobial Resistance. Msystems 2021, 6, e0118520. [Google Scholar] [CrossRef]
- Cheng, V.C.C.; Wong, S.-C.; Cao, H.; Chen, J.H.K.; So, S.Y.C.; Wong, S.C.Y.; Sridhar, S.; Yuen, K.-Y.; Ho, P.-L. Whole-genome sequencing data-based modeling for the investigation of an outbreak of community-associated methicillin-resistant Staphylococcus aureus in a neonatal intensive care unit in Hong Kong. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Kaya, H.; Hasman, H.; Larsen, J.; Stegger, M.; Johannesen, T.B.; Allesøe, R.L.; Lemvigh, C.K.; Aarestrup, F.M.; Lund, O.; Larsen, A.R. SCC mec Finder, a Web-Based Tool for Typing of Staphylococcal Cassette Chromosome mec in Staphylococcus aureus Using Whole-Genome Sequence Data. Msphere 2018, 3, e00612-17. [Google Scholar] [CrossRef] [PubMed]
- Lozano, C.; Fernández-Fernández, R.; Ruiz-Ripa, L.; Gómez, P.; Zarazaga, M.; Torres, C. Human mecC-Carrying MRSA: Clinical Implications and Risk Factors. Microorganisms 2020, 8, 1615. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef]
- Gómez-Sanz, E.; Haro-Moreno, J.M.; Jensen, S.O.; Roda-García, J.J.; López-Pérez, M. The Resistome and Mobilome of Multidrug-Resistant Staphylococcus sciuri C2865 Unveil a Transferable Trimethoprim Resistance Gene, Designated dfrE, Spread Unnoticed. Msystems 2021, 6, e0051121. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, T.; Soteyome, T.; Miao, J.; Yu, G.; Chen, D.; Ye, C.; Yang, L.; Xu, Z. Antimicrobial Resistance, SCCmec, Virulence and Genotypes of MRSA in Southern China for 7 Years: Filling the Gap of Molecular Epidemiology. Antibiotics 2023, 12, 368. [Google Scholar] [CrossRef]
- Saber, H.; Jasni, A.S.; Jamaluddin, T.Z.M.T.; Ibrahim, R. A Review of Staphylococcal Cassette Chromosome mec (SCCmec) Types in Coagulase-Negative Staphylococci (CoNS) Species. Malays. J. Med. Sci. 2017, 24, 7–18. [Google Scholar] [CrossRef]
- Maleki, A.; Pakzad, I. Association between biofilm production, adhesion genes and drugs resistance in different SCCmec types of methicillin resistant Staphylococcus aureus strains isolated from several major hospitals of Iran. Iran. J. Basic Med. Sci. 2018, 21, 400–403. [Google Scholar] [CrossRef]
- World Health Organization. Bacterial Priority Pathogens List 2024: Bacterial Pathogens of Public Health Importance, to Guide Research, Development, and Strategies to Prevent and Control Antimicrobial Resistance, 1st ed.; World Health Organization: Geneva, Switzerland, 2024. [Google Scholar]
- Abia, A.L.K.; Traore, A.N.; Potgieter, N. Editorial: Antimicrobial resistance and one health: From culture to genomics. Front. Cell. Infect. Microbiol. 2023, 13, 1294241. [Google Scholar] [CrossRef]
- Li, M.; Du, X.; Villaruz, A.E.; Diep, B.A.; Wang, D.; Song, Y.; Tian, Y.; Hu, J.; Yu, F.; Lu, Y.; et al. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat. Med. 2012, 18, 1–11. [Google Scholar] [CrossRef]
- Schroeder, K.; Jularic, M.; Horsburgh, N.; Hirschhausen, N.; Neumann, C.; Bertling, A.; Schulte, A.; Foster, S.; Kehrel, B.E.; Peters, G.; et al. Molecular characterization of a novel Staphylococcus aureus surface protein (SasC) involved in cell aggregation and biofilm accumulation. PLoS ONE 2009, 23, e7567. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | N | Clinical, N = 42,188 1 | Environment/Others, N = 1881 1 | p-Value 2 |
|---|---|---|---|---|
| Continents | 44,069 | <0.001 | ||
| Africa | 857 (2.0%) | 70 (3.7%) | ||
| Asia | 4674 (11%) | 889 (47%) | ||
| Europe | 15,957 (38%) | 408 (22%) | ||
| North America | 15,832 (38%) | 477 (25%) | ||
| Oceania | 3496 (8.3%) | 15 (0.8%) | ||
| South America | 1372 (3.3%) | 22 (1.2%) | ||
| MLST | 44,069 | <0.001 | ||
| - | 14,239 (34%) | 870 (46%) | ||
| 8 | 7501 (18%) | 162 (8.6%) | ||
| 5 | 6504 (15%) | 146 (7.8%) | ||
| 22 | 3120 (7.4%) | 11 (0.6%) | ||
| 30 | 1910 (4.5%) | 25 (1.3%) | ||
| 398 | 1527 (3.6%) | 321 (17%) | ||
| 45 | 1535 (3.6%) | 26 (1.4%) | ||
| 239 | 1472 (3.5%) | 8 (0.4%) | ||
| 1 | 1269 (3.0%) | 110 (5.8%) | ||
| 105 | 1182 (2.8%) | 15 (0.8%) | ||
| 15 | 1090 (2.6%) | 13 (0.7%) | ||
| 59 | 839 (2.0%) | 174 (9.3%) |
| Isolation Type | Number of Sequences |
| Clinical | 42,188 (96%) |
| Environmental/Other | 1881 (4.3%) |
| Total | 44,069 |
| Genes | Sequence Frequency |
| eap/map | 43,833 (99%) |
| ebps | 44,059 (100%) |
| sarA | 41,760 (95%) |
| sasG | 5635 (13%) |
| clfA1 | 43,533 (99%) |
| clfB1 | 43,906 (100%) |
| cna1 | 8557 (19%) |
| fnbA2 | 43,207 (98%) |
| fnbB1 | 35,666 (81%) |
| icaA2 | 44,013 (100%) |
| icaB | 43,854 (100%) |
| icaC1 | 43,834 (99%) |
| icaD1 | 44,005 (100%) |
| icaR2 | 43,857 (100%) |
| isdA1 | 44,059 (100%) |
| isdB1 | 44,032 (100%) |
| isdC1 | 44,060 (100%) |
| isdE1 | 44,052 (100%) |
| sasC | 43,508 (99%) |
| sdrC1 | 43,317 (98%) |
| sdrD1 | 38,972 (88%) |
| sdrE1 | 40,718 (92%) |
| ARG | Sequence Frequency |
| aac(6′)-Ie/aph(1″)-Ia | 9694 (22%) |
| abc-f | 6518 (15%) |
| ant(1″)-Ia | 11,877 (27%) |
| aph (AME) | 12,459 (28%) |
| cfr | 6435 (15%) |
| dfr | 21,895 (50%) |
| fexA-B | 20,051 (45%) |
| fusA | 12,826 (29%) |
| mecC | 15,268 (35%) |
| tet | 6786 (15%) |
| IV | 14,065 (32%) |
| mecA | 25,657 (58%) |
| mecC2 | 25,733 (58%) |
| IIa | 5500 (12%) |
| IVa | 9537 (22%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-de-Jesus, A.C.; Ferrari, R.G.; Panzenhagen, P.; dos Santos, A.M.P.; Portes, A.B.; Conte-Junior, C.A. Distribution of Antimicrobial Resistance and Biofilm Production Genes in the Genomic Sequences of S. aureus: A Global In Silico Analysis. Antibiotics 2025, 14, 364. https://doi.org/10.3390/antibiotics14040364
Silva-de-Jesus AC, Ferrari RG, Panzenhagen P, dos Santos AMP, Portes AB, Conte-Junior CA. Distribution of Antimicrobial Resistance and Biofilm Production Genes in the Genomic Sequences of S. aureus: A Global In Silico Analysis. Antibiotics. 2025; 14(4):364. https://doi.org/10.3390/antibiotics14040364
Chicago/Turabian StyleSilva-de-Jesus, Ana Carolina, Rafaela G. Ferrari, Pedro Panzenhagen, Anamaria M. P. dos Santos, Ana Beatriz Portes, and Carlos Adam Conte-Junior. 2025. "Distribution of Antimicrobial Resistance and Biofilm Production Genes in the Genomic Sequences of S. aureus: A Global In Silico Analysis" Antibiotics 14, no. 4: 364. https://doi.org/10.3390/antibiotics14040364
APA StyleSilva-de-Jesus, A. C., Ferrari, R. G., Panzenhagen, P., dos Santos, A. M. P., Portes, A. B., & Conte-Junior, C. A. (2025). Distribution of Antimicrobial Resistance and Biofilm Production Genes in the Genomic Sequences of S. aureus: A Global In Silico Analysis. Antibiotics, 14(4), 364. https://doi.org/10.3390/antibiotics14040364