Antimicrobial Action and Reversal of Resistance in MRSA by Difluorobenzamide Derivatives Targeted at FtsZ

 , , , , and
, , , , and 
Abstract
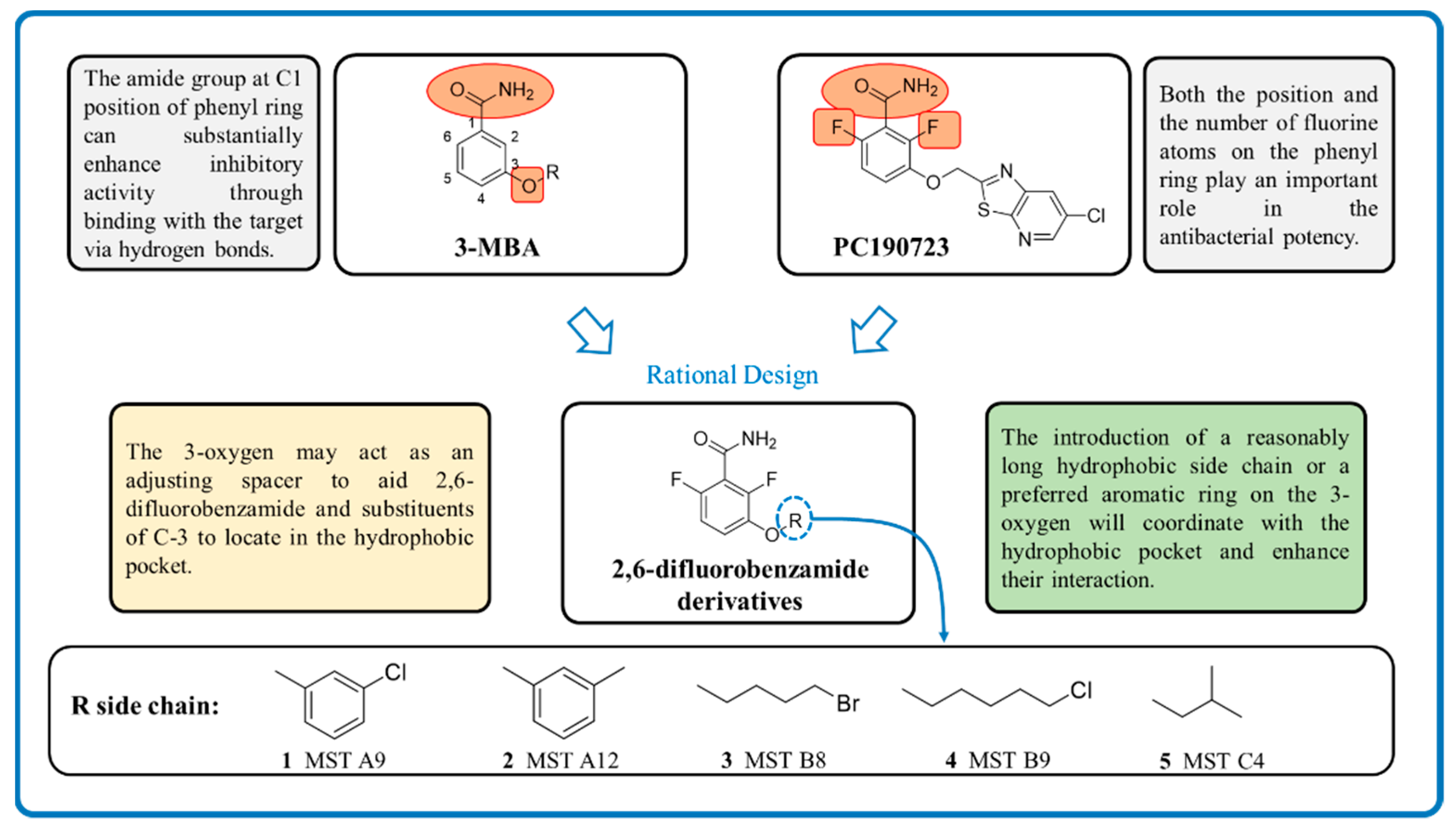
1. Introduction
2. Results and Discussion
2.1. Antimicrobial Activity of MST Compounds
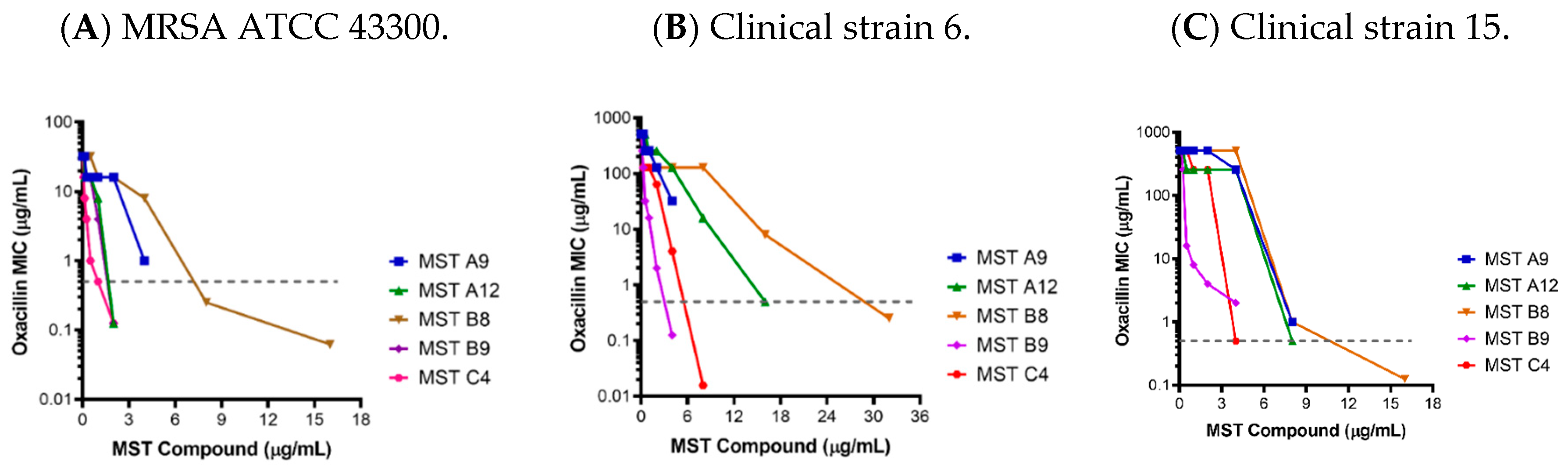
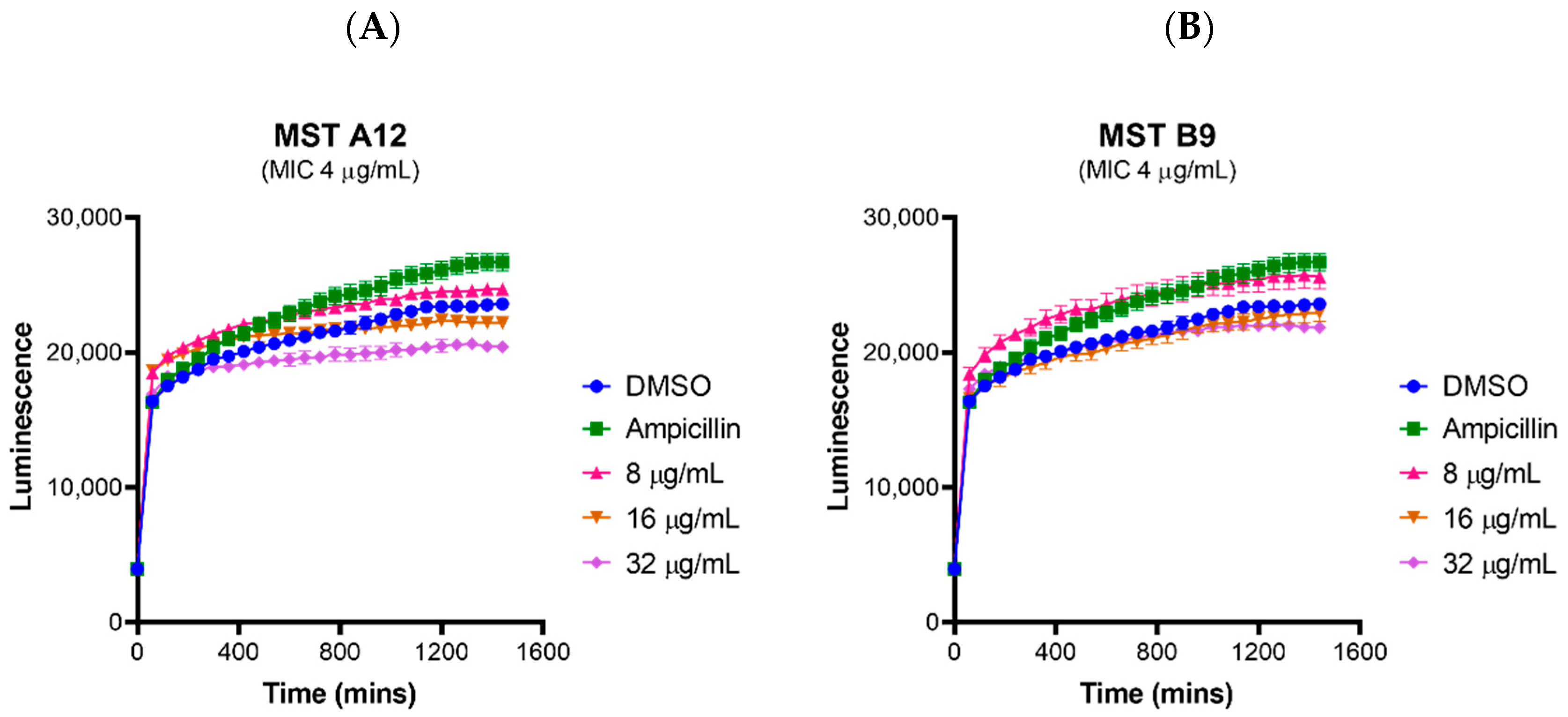
2.2. MST Compounds Reverse Resistance to Oxacillin in MRSA ATCC 43300 and Clinical MRSA Isolates
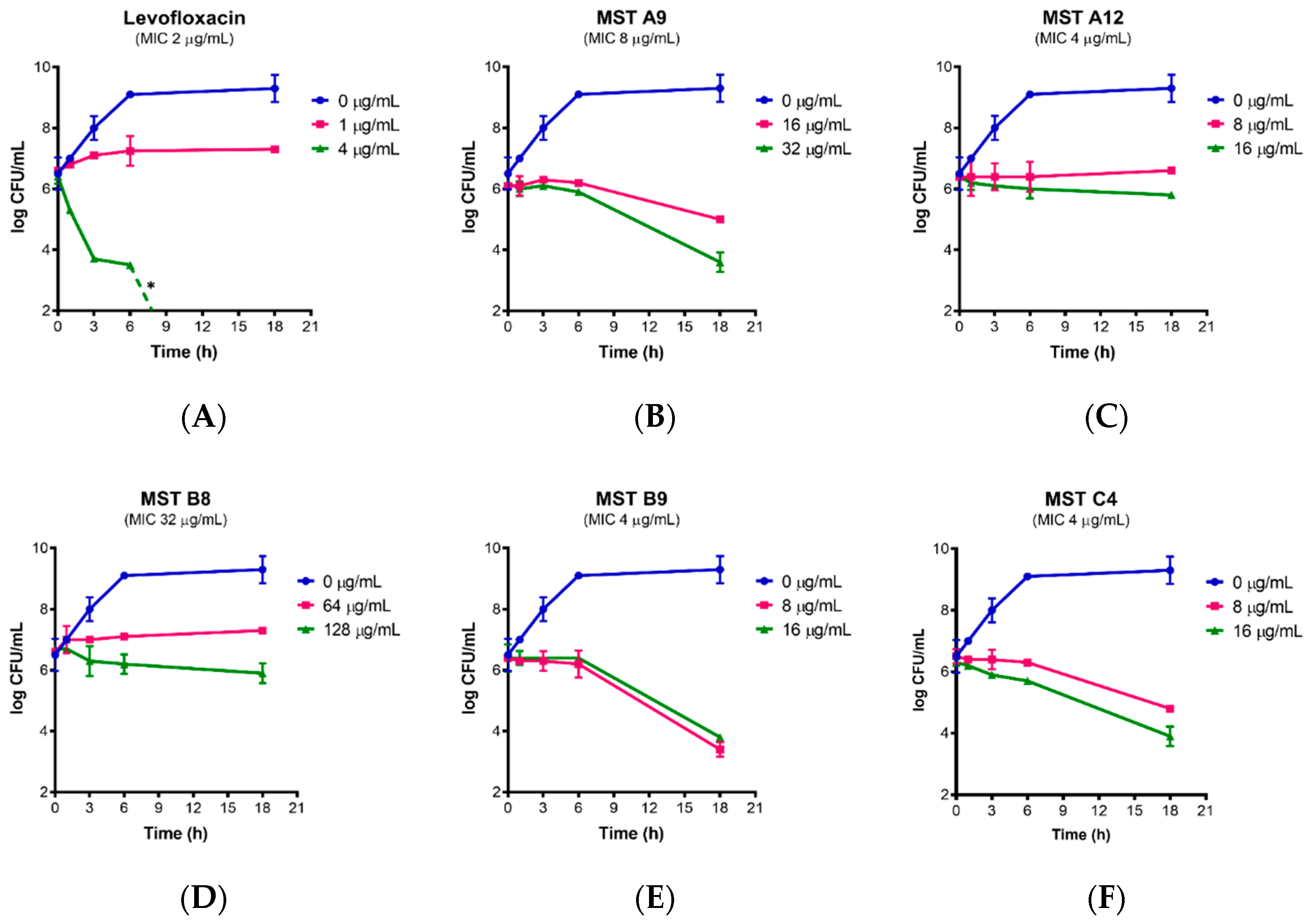
2.3. Time Kill Curves for MST Compounds Indicate Bactericidal or Bacteriostatic Mechanisms
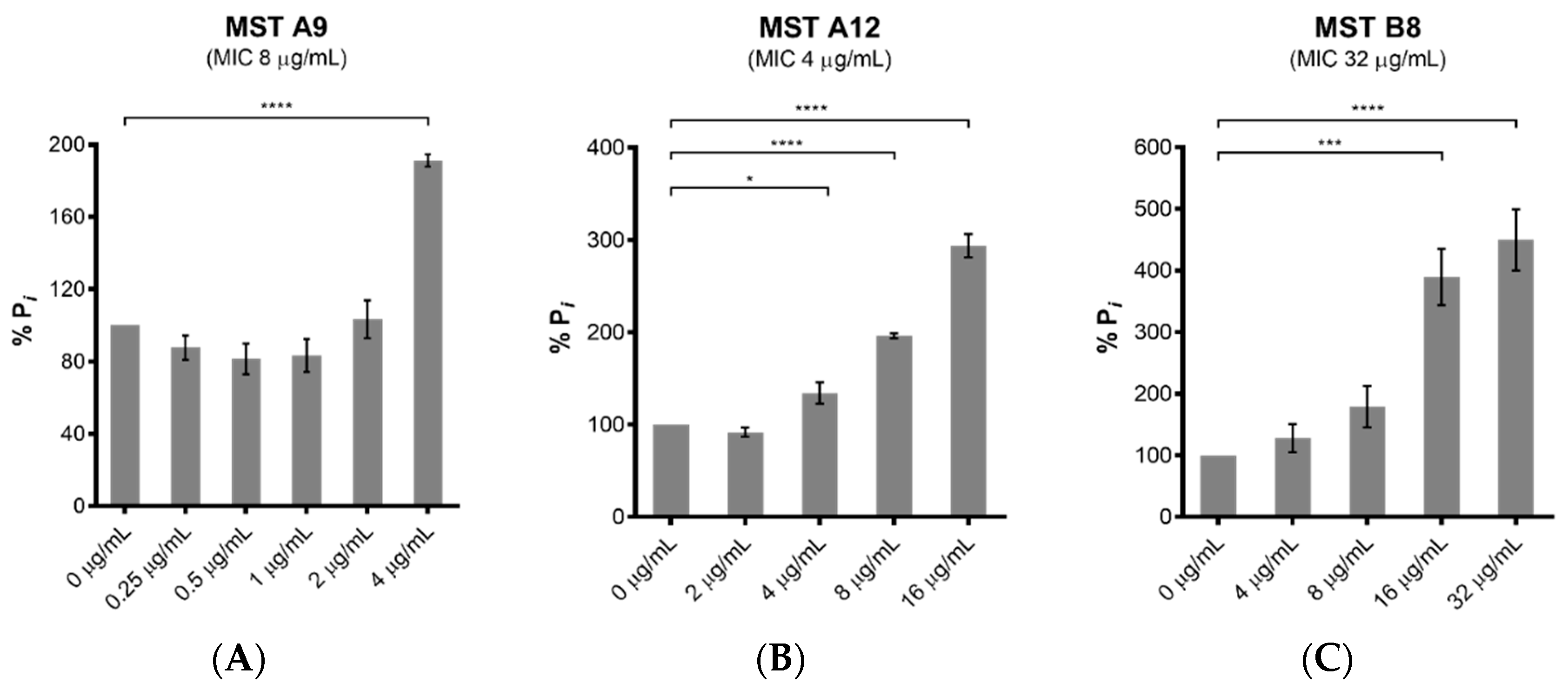
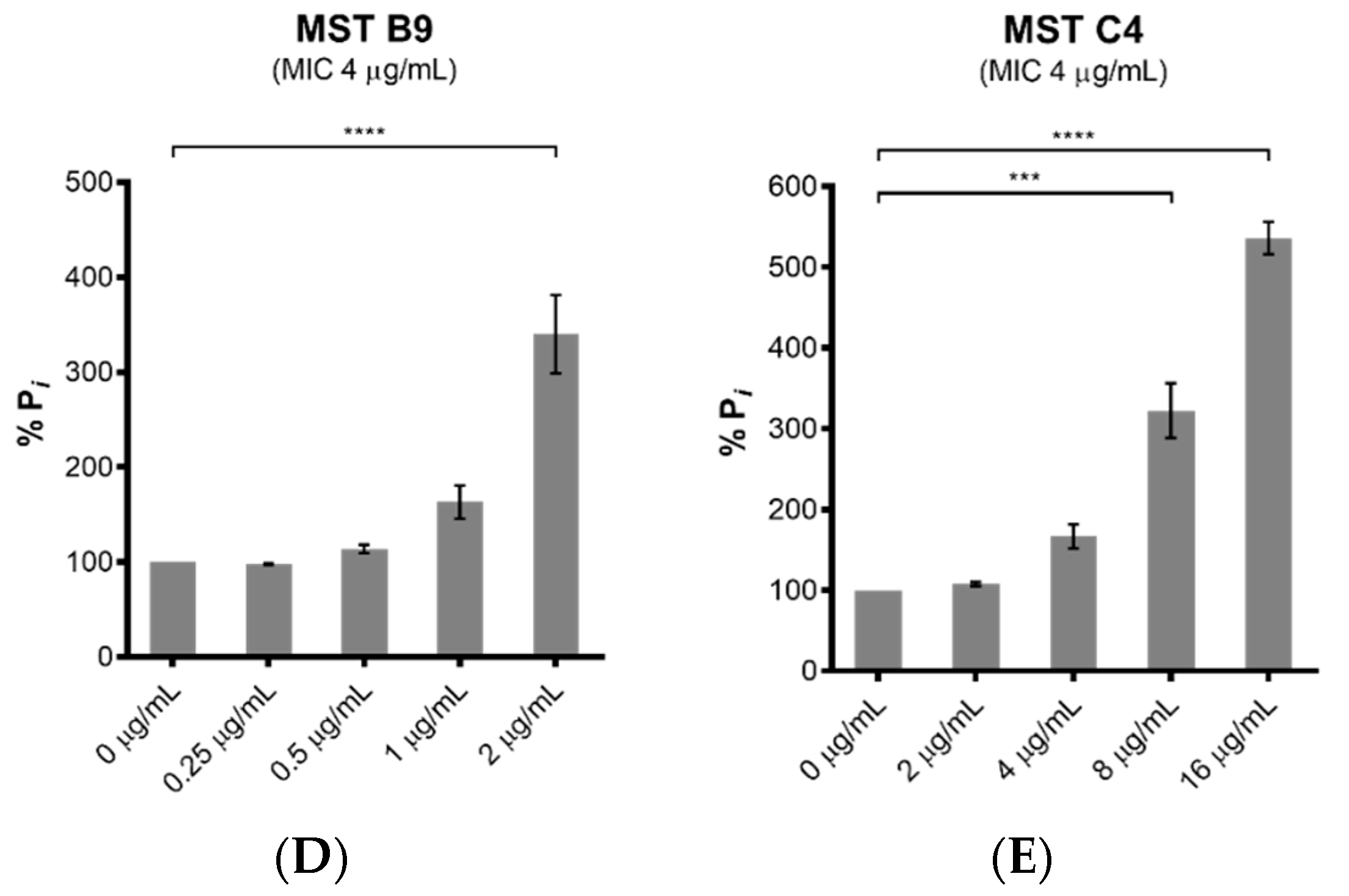
2.4. MST Compounds Disrupt Cellular Division
2.5. Preparation of Recombinant FtsZ from Staphylococcus Aureus
2.6. MST Compounds Enhance SaFtsZ GTPase Activity
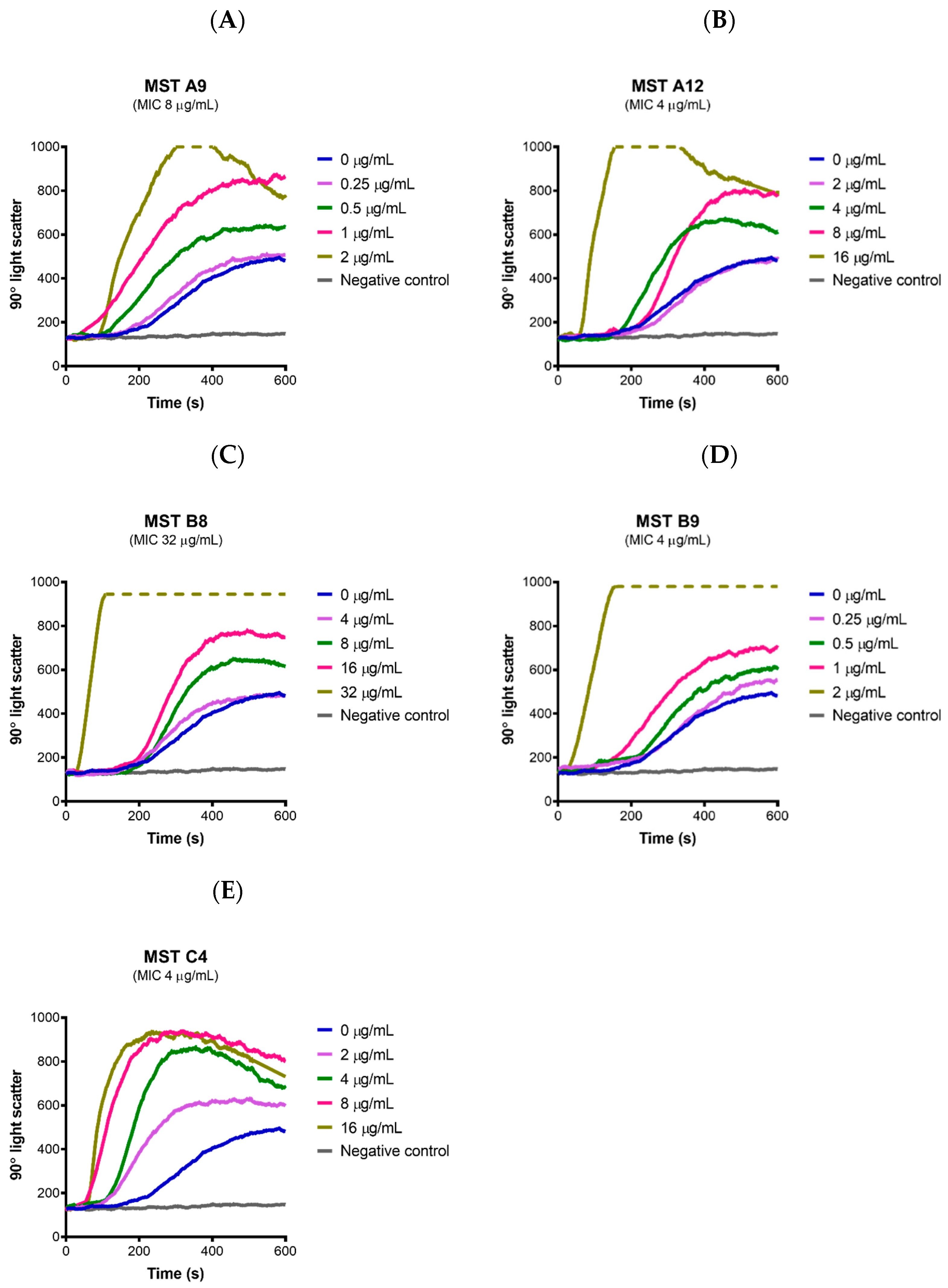
2.7. MST Compounds Stabilize SaFtsZ Polymerization
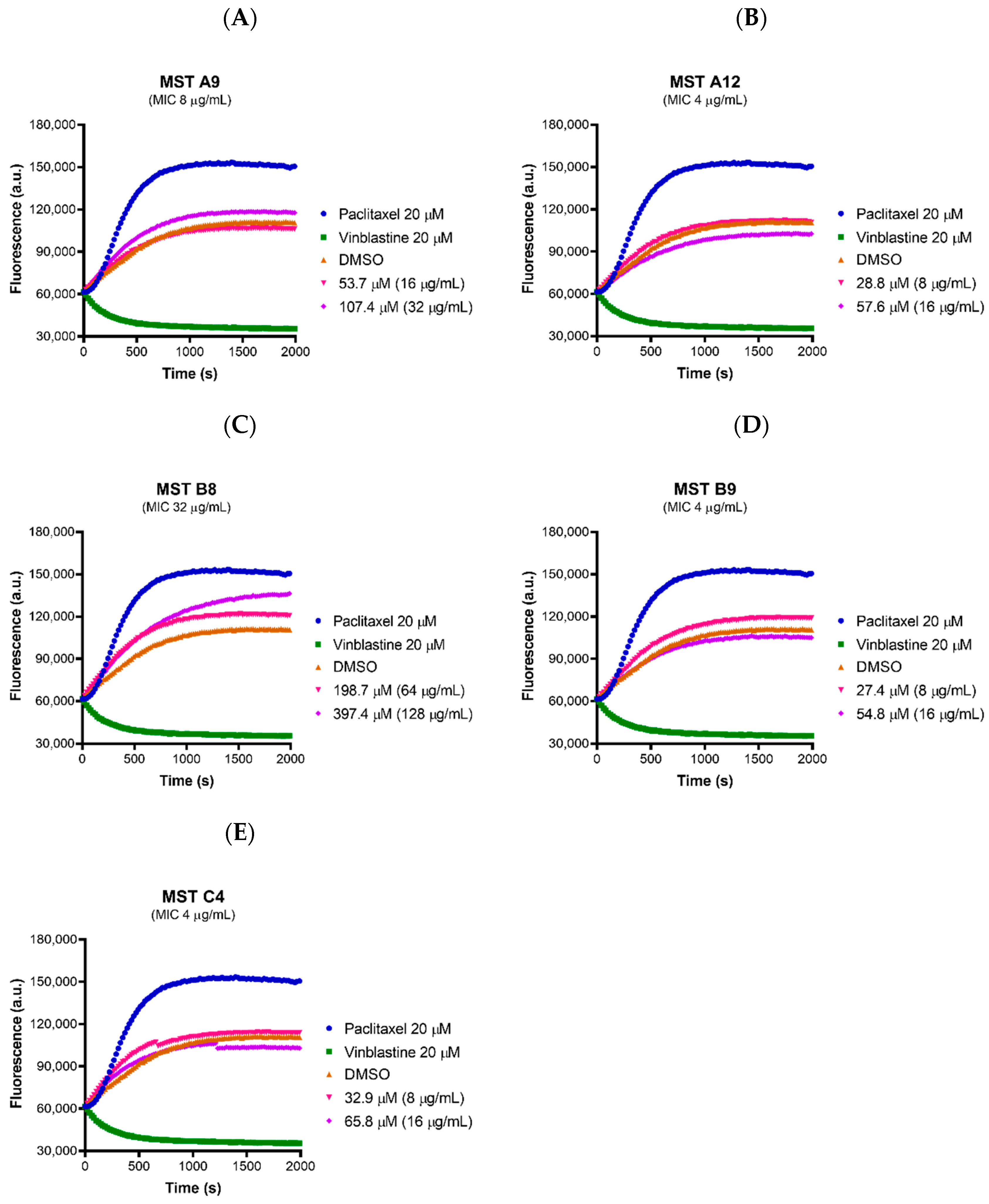
2.8. MST Compounds Do Not Affect the Polymerization of Mammalian Tubulin
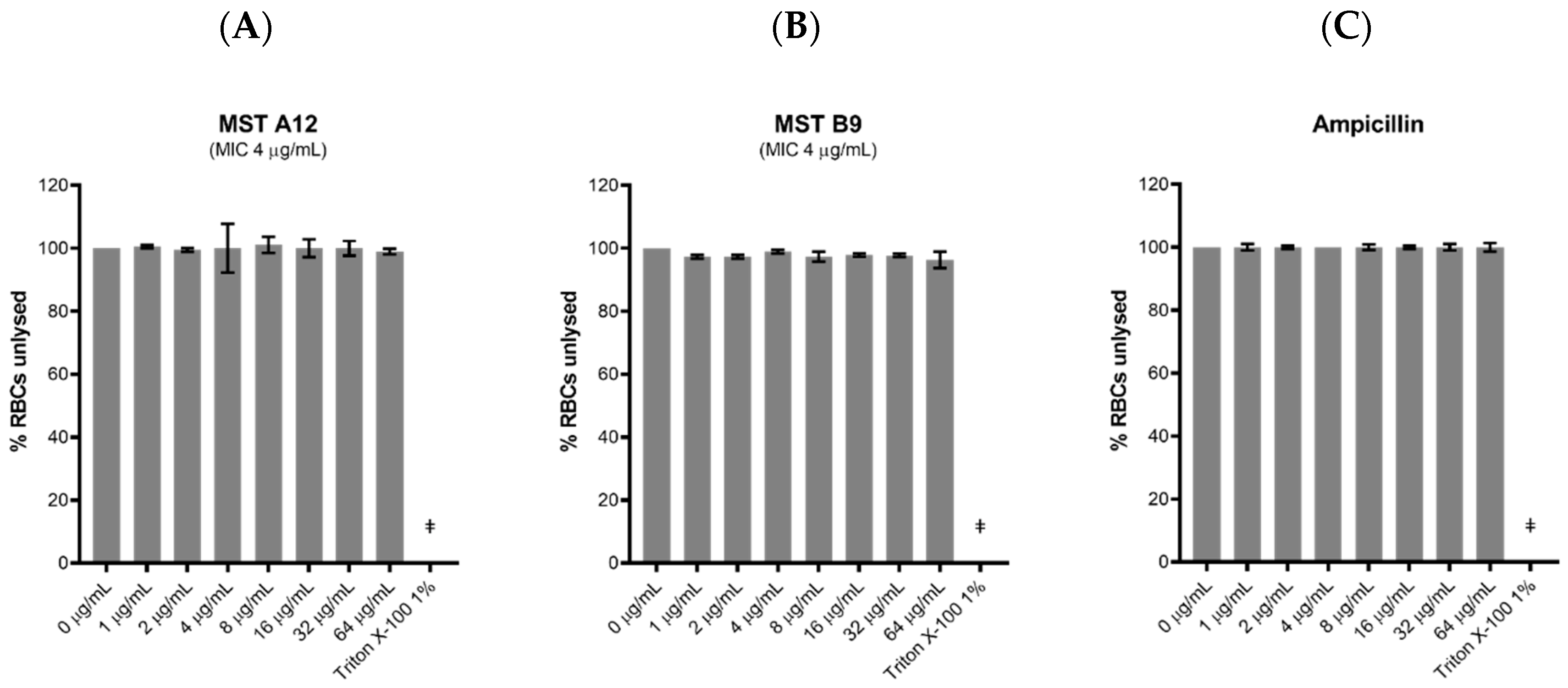
2.9. Toxicity of the MST Compounds
3. Materials and Methods
3.1. Chemicals
3.2. Synthesis of Compounds
3.3. Bacterial Strains and Growth Conditions
3.4. Drug Susceptibility Assay
3.5. Synergistic Activity between Oxacillin and Benzamide Derivatives on MRSA
3.6. Time Kill Assays against MRSA
3.7. Determination of Cell Division Phenotype
3.8. Cloning of MRSA ATCC 43300 FtsZ Protein
3.9. Over-Expression and Purification of FtsZ Protein
3.10. Protein Concentration Determination
3.11. GTPase Assay
3.12. Effects on SaFtsZ Polymerization Using 90° Light Scattering
3.13. Mammalian Tubulin Polymerization Assay
3.14. Cytotoxicity Analysis of MST Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling drug-resistant Infections Globally, Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.; Sumpradit, N.; Vleieghe, E.; Hara, G.L.; Gould, I.M.; Goosses, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- World Health Organization. Global Action Plan on Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Kusuma, K.D.; Payne, M.; Ung, A.T.; Bottomley, A.L.; Harry, E.J. FtsZ as an antibacterial target, Status and guidelines for progressing this avenue. ACS Infect. Dis. 2019, 5, 1279–1294. [Google Scholar] [CrossRef] [PubMed]
- Bisson-Filho, A.W.; Hsu, Y.-P.; Squyres, G.R.; Kuru, E.; Wu, F.; Jukes, C.; Sun, Y.; Dekker, C.; Holden, S.; VanNieuwenhze, M.S.; et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 2017, 355, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Löwe, J.; Amos, L.A. Crystal structure of the bacterial cell-division protein FtsZ. Nature 1998, 391, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Yamane, J.; Mogi, N.; Yamaguchi, H.; Takemoto, H.; Yao, M.; Tanaka, I. Structural reorganization of the bacterial cell-division protein FtsZ from Staphylococcus aureus. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Huecas, S.; Ramírez-Aportela, E.; Vergoñós, A.; Núñez-Ramírez, R.; Llorca1, O.; Díaz, J.F.; Juan-Rodriguez, D.; Olivia, M.A.; Castellen, P.; Andreu, J.M. Self-organization of FtsZ polymers in solution reveals spacer role of the disordered C-terminal tail. Boiophys. J. 2017, 113, 1831–1844. [Google Scholar] [CrossRef]
- Nogales, E.; Downing, K.H.; Amos, L.A.; Löwe, J. Tubulin and FtsZ form a distinct family of GTPases. Nat. Struct. Biol. 1998, 5, 451–458. [Google Scholar] [CrossRef]
- Kusuma, K.D.; Griffith, R.; Harry, E.J.; Bottomley, A.L.; Ung, A.T. In silico analysis of FtsZ crystal structures towards a new target for antibiotics. Aust. J. Chem. 2019, 72, 184–193. [Google Scholar] [CrossRef]
- Carro, L. Recent progress in the development of small-molecule FtsZ inhibitors as chemical tools for the development of novel antibiotics. Antibiotics 2019, 8, 217. [Google Scholar] [CrossRef]
- Casiraghi, A.; Suigo, L.; Valoti, E.; Straniero, V. Targeting bacterial cell division, A binding site-centered approach to the most promising inhibitors of the essential protein FtsZ. Antibiotics 2020, 9, 69. [Google Scholar] [CrossRef]
- Kapoor, S.; Panda, D. Targeting FtsZ for antibacterial therapy, A promising avenue. Expert Opin. Ther. Targets 2009, 13, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Mark, L.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. Combining the FtsZ-targeting prodrug TXA709 and the cephalosporin cefdinir confers synergy and reduces the frequency of resistance in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2016, 60, 4290–4296. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.G.; Collins, I.; Boyd, E.A.; Brown, D.; East, S.P.; Gardiner, M.; Fletcher, R.; Haydon, D.J.; Henstock, V.; Ingram, P.; et al. Antibacterial alkoxybenzamide inhibitors of the essential bacterial cell division protein FtsZ. Bioorg. Med. Chem. Lett. 2009, 19, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Stokes, N.R.; Baker, N.; Bennett, J.M.; Chauhan, P.K.; Collins, I.; Davies, D.T.; Gavade, M.; Kumar, D.; Lancett, P.; Macdonald, R.; et al. Design, synthesis and structure-activity relationships of substituted oxazole-benzamide antibacterial inhibitors of FtsZ. Bioorg. Med. Chem. Lett. 2014, 24, 353–359. [Google Scholar] [CrossRef]
- Chiodini, G.; Pallavicini, M.; Zanotto, C.; Bissa, M.; Radaelli, A.; Straniero, V.; Bolchi, C.; Fumagalli, L.; Ruggeri, P.; Morghen, C.D.G.; et al. Benzodioxane-benzamides as new bacterial cell division inhibitors. Eur. J. Med. Chem. 2015, 89, 252–265. [Google Scholar] [CrossRef]
- Andreu, J.M.; Schaffner-Barbero, C.; Huecas, S.; Alonso, D.; Lopez-Rodriguez, M.L.; Ruiz-Avila, L.B.; Núñez-Ramírez, R.; Llorca, O.; Martín-Galiano, A.J. The antibacterial cell division inhibitor PC190723 is an FtsZ polymer-stabilizing agent that induces filament assembly and condensation. J. Biol. Chem. 2010, 285, 14239–14246. [Google Scholar] [CrossRef]
- World Health Organization. Antibacterial Agents in Clinical Development, An Analysis of the Antibacterial Clinical Development Pipeline; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Kaul, M.; Zhang, Y.; Parhi, A.K.; Lavoie, E.J.; Pilch, D.S. Inhibition of RND-type efflux pumps confers the FtsZ-directed prodrug TXY436 with activity against Gram-negative bacteria. Biochem. Pharmacol. 2014, 89, 321–328. [Google Scholar] [CrossRef]
- Straniero, V.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Casiraghi, A.; Suigo, L.; Zdovc, I.; Radaelli, A.; De Giuli Morghen, C.; Valoti, E. Benzodioxane-benzamides as antibacterial agents, Computational and SAR studies to evaluate the influence of the 7-Substitution in FtsZ Interaction. ChemMedChem 2020, 15, 195–209. [Google Scholar] [CrossRef]
- Straniero, V.; Suigo, L.; Casiraghi, A.; Sebastián-Pérez, V.; Hrast, M.; Zanotto, C.; Zdovc, I.; De Giuli Morghen, C.; Radaelli, A.; Valoti, E. Benzamide derivatives targeting the cell division protein FtsZ, Modifications of the linker and the benzodioxane scaffold and their effects on antimicrobial activity. Antibiotics 2020, 9, 160. [Google Scholar] [CrossRef]
- Qiang, S.; Wang, C.; Venter, H.; Li, X.; Wang, Y.; Guo, L.; Ma, R.; Ma, S. Synthesis and biological evaluation of novel FtsZ-targeted 3-arylalkoxy-2,6-difluorobenzamides as potential antimicrobial agents. Chem. Biol. Drug Des. 2016, 87, 257–264. [Google Scholar] [CrossRef]
- Bi, F.; Guo, L.; Wang, Y.; Venter, H.; Semple, S.J.; Liu, F.; Ma, S. Design, synthesis and biological activity evaluation of novel 2,6-difluorobenzamide derivatives through FtsZ inhibition. Bioorg. Med. Chem. Lett. 2017, 27, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens, No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Priority Pathogens List for Research and Development of New Antibiotics; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Venter, H. Reversing resistance to counter antimicrobial resistance in the World Health Organisation’s critical priority of most dangerous pathogens. Biosci. Rep. 2019, 39, BSR0180474. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.M.; Therien, A.G.; Lu, J.; Lee, S.H.; Caron, A.; Gill, C.J.; Lebeau-Jacob, C.; Benton-Perdomo, L.; Monteiro, J.M.; Pereira, P.M.; et al. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-Lactam antibiotics. Sci. Transl. Med. 2012, 4, 126ra35. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Tateda, K.; Kikuchi, Y.; Kimura, S.; Miyazaki, C.; Ishii, Y.; Tanabe, Y.; Geyjo, F.; Yamaguchi, K. Efficacy of colistin combination therapy in a mouse model of pneumonia caused by multidrug-resistant Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2009, 63, 534–542. [Google Scholar] [CrossRef]
- Wang, Y.; Alenzy, R.; Song, D.; Liu, X.; Teng, Y.; Mowla, R.; Ma, Y.; Polyak, S.W.; Venter, H.; Ma, S. Structural optimization of natural product nordihydroguaretic acid to discover novel analogues as AcrB inhibitors. Eur. J. Med. Chem. 2020, 186, 111910. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Du, R.L.; Cai, S.Y.; Liu, Z.H.; Fang, Z.Y.; Liu, T.; So, L.Y.; Lu, Y.J.; Sun, N.; Wong, K.Y. Study of benzofuroquinolinium derivatives as a new class of potent antibacterial agent and the mode of inhibition targeting FtsZ. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Fang, Z.; Ban, L.; Li, Y.; Yuan, W.; Liu, Z.; Liu, T.; Li, X.; Wong, K.Y.; Lu, Y.; Sun, N.; et al. A quinoline-based FtsZ inhibitor for the study of antimicrobial activity and synergistic effects with β-lactam antibiotics. J. Pharmacol. Sci. 2018, 137, 283–289. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, W.; Zhu, Y.; Shi, S.; Li, Q.; Mao, C.; Zhao, D.; Zhan, Y.; Shi, J.; Li, W.; et al. Inhibiting methicillin-resistant Staphylococcus aureus by tetrahedral DNA nanostructure-enabled antisense peptide nucleic acid delivery. Nano Lett. 2018, 18, 5652–5659. [Google Scholar] [CrossRef]
- Haydon, D.J.; Stokes, N.R.; Ure, R.; Galbraith, G.; Bennett, J.M.; Brown, D.R. An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 2008, 321, 1673–1675. [Google Scholar] [CrossRef]
- Bi, F.; Song, D.; Zhang, N.; Liu, Z.; Gu, X.; Hu, C.; Cai, X.; Venter, H.; Ma, S. Design, synthesis and structure-based optimization of novel isoxazole-containing benzamide derivatives as FtsZ modulators. Eur. J. Med. Chem. 2018, 159, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. An FtsZ-targeting prodrug with oral antistaphylococcal efficacy in vivo. Antimicrob. Agents Chemother. 2013, 57, 5860–5869. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; Lyu, Y.L.; Pawlak, J.; Saravolatz, S.; Saravolatz, L.D.; Weinstein, M.P.; LaVoie, E.J.; et al. TXA709, an FtsZ-targeting benzamide prodrug with improved pharmacokinetics and enhanced in vivo efficacy against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 4845–4855. [Google Scholar] [CrossRef] [PubMed]
- Annapurna, S.M.; Harikrishna, S.S.; Aravind, K.B.; Pawar, P.D. Targeting FtsZ as novel therapeutic strategy against multi-drug resistant Staphylococcus aureus. Asian J. Microbiol. Biotechnol. Environ. Sci. 2015, 17, 131–138. [Google Scholar]
- Sun, N.; Lu, Y.J.; Chan, F.Y.; Du, R.L.; Zheng, Y.; Zhang, K.; So, L.Y.; Abagyan, R.; Zhuo, C.; Leung, Y.C.; et al. A thiazole orange derivative targeting the bacterial protein FtsZ shows potent antibacterial activity. Front. Microbiol. 2017, 8, 855. [Google Scholar] [CrossRef]
- Adams, D.W.; Wu, L.J.; Errington, J. A benzamide-dependent FtsZ mutant reveals residues crucial for Z-ring assembly. Mol. Microbiol. 2016, 99, 1028–1042. [Google Scholar] [CrossRef]
- Straniero, V.; Zanotto, C.; Straniero, L.; Casiraghi, A.; Duga, S.; Radaelli, A.; De Giuli Morghen, C.; Valoti, E. 2,6-difluorobenzamide inhibitors of bacterial cell division protein FtsZ, Design, synthesis, and structure-activity relationships. ChemMedChem 2017, 12, 1303–1318. [Google Scholar] [CrossRef]
- Elsen, N.L.; Lu, J.; Parthasarathy, G.; Reid, J.C.; Sharma, S.; Soisson, S.M.; Lumb, K.J. Mechanism of action of the cell-division inhibitor PC190723, Modulation of FtsZ assembly cooperativity. J. Am. Chem. Soc. 2012, 134, 12342–12345. [Google Scholar] [CrossRef]
- Stokes, N.R.; Sievers, J.; Barker, S.; Bennett, J.M.; Brown, D.R.; Collins, I.; Errington, V.M.; Foulger, D.; Hall, M.; Halsey, R.; et al. Novel inhibitors of bacterial cytokinesis identified by a cell-based antibiotic screening assay. J. Biol. Chem. 2005, 280, 39709–39715. [Google Scholar] [CrossRef]
- Kaul, M.; Parhi, A.K.; Zhang, Y.; Lavoie, E.J.; Tuske, S.; Arnold, E.; Kerrigan, J.E.; Pilch, D.S. A bactericidal guanidinomethyl biaryl that alters the dynamics of bacterial FtsZ polymerization. J. Med. Chem. 2012, 55, 10160–10176. [Google Scholar] [CrossRef]
- Löwe, J.; Li, H.; Downing, K.H.; Nogales, E. Refined structure of αβ-tubulin at 3.5 Å resolution. J. Mol. Biol. 2001, 313, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Du, R.L.; Zheng, Y.Y.; Guo, Q.; Cai, S.Y.; Liu, Z.H.; Fang, Z.Y.; Yuan, W.C.; Liu, T.; Li, X.M.; et al. Antibacterial activity of 3-methylbenzo[d]thiazol-methylquinolinium derivatives and study of their action mechanism. J. Enzym. Inhib. Med. 2018, 33, 879–889. [Google Scholar] [CrossRef]
- Sun, N.; Zheng, Y.Y.; Du, R.L.; Cai, S.Y.; Zhang, K.; So, L.Y.; Cheung, K.C.; Zhuo, C.; Lu, Y.J.; Wong, K.Y. New application of tiplaxtinin as an effective FtsZ-targeting chemotype for an antimicrobial study. MedChemComm 2017, 8, 1909–1913. [Google Scholar] [CrossRef] [PubMed]
- Hurley, K.A.; Santos, T.M.A.; Nepomuceno, G.M.; Huynh, V.; Shaw, J.T.; Weibel, D.B. Targeting the bacterial division protein FtsZ. J. Med. Chem. 2016, 59, 6975–6998. [Google Scholar] [CrossRef] [PubMed]
- Coombs, G.W.; Pearson, J.C.; Christiansen, K.J.; Nimmo, G.R. Staphylococcus aureus Programme 2010 (SAP 2010) Community Survey MRSA Epidemiology and Typing Report; The Australian Group on Antimicrobial Resistance: Sydney, New South Wales, Australia, 2010. [Google Scholar]
- Welch, A.; Awah, C.U.; Jing, S.H.; Van Veen, H.W.; Venter, H. Promiscuous partnering and independent activity of MexB, the multidrug transporter protein from Pseudomonas aeruginosa. Biochem. J. 2010, 430, 355–364. [Google Scholar] [CrossRef]
- European Committee for Antimicrobial Susceptibility Testing. Media for MIC Determination by the Broth Microdilution Method; European Committee for Antimicrobial Susceptibility Testing: Vaxjo, Sweden, 2016. [Google Scholar]
- Doern, C.D. When does 2 plus 2 equal 5? A review of antimicrobial synergy testing. J. Clin. Microbiol. 2014, 52, 4124–4128. [Google Scholar] [CrossRef]
- Seyedmohammad, S.; Fuentealba, N.A.; Marriott, R.A.J.; Goetze, T.A.; Edwardson, J.M.; Barrera, N.P.; Venter, H. Structural model of FeoB, the iron transporter from Pseudomonas aeruginosa, predicts a cysteine lined, GTP-gated pore. Biosci. Rep. 2016, 36, 1–25. [Google Scholar] [CrossRef]
- Wang, Y.; Mowla, R.; Guo, L.; Ogunniyi, A.D.; Rahman, T.; De Barros Lopes, M.A.; Ma, S.; Venter, H. Evaluation of a series of 2-napthamide derivatives as inhibitors of the drug efflux pump AcrB for the reversal of antimicrobial resistance. Bioorg. Med. Chem. Lett. 2017, 27, 733–739. [Google Scholar] [CrossRef]
- Porta de la Riva, M.; Fontrodona, L.; Villanueva, A.; Ceron, J. Basic Caenorhabditis elegans methods, synchronization and observation. J. Vis. Exp. 2012, 64, e4019. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compounds and Antimicrobial Agents | Minimum Inhibitory Concentration (MIC), µg/mL | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ATCC Strains | Clinical MRSA Strains | VRE WW 734 | ||||||||||||||||||
| Compound | R side chain | MSSAATCC 25923 | MRSAATCC 43300 | C1 | C2 | C3 | C6 | C7 | C8 | C9 | C10 | C11 | C13 | C14 | C15 | C16 | C18 | C19 | C20 | |
| 1 MST A9 |  | 4 | 8 | 16 | 16 | 16 | 32 | 16 | 8 | 8 | 8 | 8 | 16 | 16 | 16 | 16 | 16 | 16 | 16 | >64 |
| 3-((3-chlorobenzyl)oxy)-2,6-difluorobenzamide | ||||||||||||||||||||
| 2 MST A12 |  | 4 | 4 | 16 | 16 | 16 | 16 | 16 | 16 | 16 | 16 | 32 | 16 | 16 | 16 | 16 | 16 | 16 | 16 | >64 |
| 3-((3-methylbenzyl)oxy)-2,6-difluorobenzamide | ||||||||||||||||||||
| 3 MST B8 |  | 8 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | 64 | 64 | 32 | 32 | 32 | 32 | 32 | 32 | 32 | >64 |
| 3-((5-bromopentyl)oxy)-2,6-difluorobenzamide | ||||||||||||||||||||
| 4 MST B9 |  | 1 | 4 | 8 | 16 | 8 | 4 | 4 | 8 | 4 | 4 | 8 | 4 | 8 | 8 | 8 | 8 | 8 | 4 | >64 |
| 3-((6-chlorohexyl)oxy)-2,6-difluorobenzamide | ||||||||||||||||||||
| 5 MST C4 |  | 2 | 4 | 8 | 8 | 8 | 8 | 8 | 16 | 8 | 16 | 16 | 8 | 16 | 8 | 8 | 16 | 8 | 8 | 64 |
| 3-(isopentyloxy)-2,6-difluorobenzamide | ||||||||||||||||||||
| Divin | >128 | >128 | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | n.t. | |
| Oxacillin | 0.5 | 32 | 64 | 64 | 32 | 512 | 256 | 32 | 256 | 256 | 512 | 128 | 512 | 512 | 32 | 64 | 64 | 32 | >512 | |
| Levofloxacin | 0.5 | 2 | 0.25 | 64 | 0.25 | 32 | 16 | 0.25 | 32 | 16 | 8 | 16 | 8 | 0.25 | 16 | 0.25 | 0.5 | 0.25 | >64 | |
| Vancomycin | n.t. | 0.5 | 1 | 1 | 1 | 1 | 0.5 | 1 | 0.5 | 1 | 1 | 1 | 1 | 4 | 1 | 1 | 1 | 1 | 64 | |
| Compounds | MIC, µg/mL | |
|---|---|---|
| Colistin | ||
| +0 µg/mL | +0.125 µg/mL | |
| 1 MST A9 | >256 | >256 |
| 2 MST A12 | >256 | 128 |
| 3 MST B8 | >256 | >256 |
| 4 MST B9 | >256 | 64 |
| 5 MST C4 | >256 | 64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, W.C.; Whittall, J.J.; Song, D.; Polyak, S.W.; Ogunniyi, A.D.; Wang, Y.; Bi, F.; Ma, S.; Semple, S.J.; Venter, H. Antimicrobial Action and Reversal of Resistance in MRSA by Difluorobenzamide Derivatives Targeted at FtsZ. Antibiotics 2020, 9, 873. https://doi.org/10.3390/antibiotics9120873
Chai WC, Whittall JJ, Song D, Polyak SW, Ogunniyi AD, Wang Y, Bi F, Ma S, Semple SJ, Venter H. Antimicrobial Action and Reversal of Resistance in MRSA by Difluorobenzamide Derivatives Targeted at FtsZ. Antibiotics. 2020; 9(12):873. https://doi.org/10.3390/antibiotics9120873
Chicago/Turabian StyleChai, Wern Chern, Jonathan J. Whittall, Di Song, Steven W. Polyak, Abiodun D. Ogunniyi, Yinhu Wang, Fangchao Bi, Shutao Ma, Susan J. Semple, and Henrietta Venter. 2020. "Antimicrobial Action and Reversal of Resistance in MRSA by Difluorobenzamide Derivatives Targeted at FtsZ" Antibiotics 9, no. 12: 873. https://doi.org/10.3390/antibiotics9120873
APA StyleChai, W. C., Whittall, J. J., Song, D., Polyak, S. W., Ogunniyi, A. D., Wang, Y., Bi, F., Ma, S., Semple, S. J., & Venter, H. (2020). Antimicrobial Action and Reversal of Resistance in MRSA by Difluorobenzamide Derivatives Targeted at FtsZ. Antibiotics, 9(12), 873. https://doi.org/10.3390/antibiotics9120873