Abstract
A Stanene, is a two-dimensional material composed of tin atoms arranged in a single hexagonal layer, in a manner similar to graphene. First principle studies based on density functional theory were performed to investigate the effects of hydrogenation and Al/P doping on electronic structure and magnetic properties of stanene. Hydrogenation opens the bandgap of stanene and changes it from nonmagnetic to the ferromagnetic material through H 1s states and Sn 5p states hybridization. Al/P atom at hollow site prevent electrons of adjacent Sn atoms to connect so that inducing unpaired electrons. The combination of hydrogenation and Al/P doping increases its magnetization. The sequence based on its magnetic moment from small to large is as follows: pure stanene, Al-doped stanene, P-doped stanene, hydrogenated stanene, Al-doped hydrogenated stanene, and P-doped hydrogenated stanene. The controllable transformation from nonmagnetic metallic to a magnetic semiconductor is a key requirement for materials to be used as spintronic materials. Thus, these results may shed light on designing the stanene-based electronic and spintronics materials.
1. Introduction
Stanene, the tin analog of graphene, is one of the two-dimensional graphene-like materials that have attracted tremendous interest on account of their exciting electronic, mechanical, chemical, and physical properties and various potential applications. Among two-dimensional materials with honeycomb structure, stanene has special performance and potentials such as large gap Quantum Spin Hall (QSH) insulator [1,2], low lattice thermal conductivity which indicates higher thermoelectric [3], a good candidate for a 2d topological superconductor [4], and giant magnetoresistance [5].
Unlike graphene which is flat, germanene, silicene, and stanene were predicted theoretically and proved experimentally buckled [6]. It is believed that the buckle leads to deviations away from a sp2 hybridization. However, recently ultra-flat (zero-buckling) stanene could be grown on Cu substrate [7,8] and Ag substrate [9].
Besides the superior and unique properties of 2D honeycomb crystalline structure materials, their zero-gap makes them difficult to apply in electronic devices such as high-performance field-effect transistors (FETs) operating at room temperature. Therefore, opening a sizable band gap is one of the key issues in present research on them. Several attempts have been made to tune the electronic and magnetic behavior of 2D honeycomb crystal structure materials such as applying strain and external electric field, as well as functionalization with foreign adatoms. The effects of electric field and strain on the electronic properties of stanene have been predicted by several theoretical works [10,11], and confirmed by Jian Guo et al. using a home-built ultrahigh vacuum STM/MBE system [12]. Previous studies also reported that functionalization with foreign adatoms provided several possible routes to tune the electronic and magnetic properties [13]. Previous first principle studies suggested that the elements of alkali metals (Li−K), alkaline earth metals (Be−Ca), transition metal (Sc−Zn), and group III−VII atoms such as metals (Al, Ga), metalloids (B, Si, Ge, As) and non-metals (C, N, O, P, S, Se, F, Cl, Br) on stanene can tune their electronic properties and lead to the formation of dumbbell structures [14]. Hydrogenation of stanene also can change the electronic and magnetic properties of stanene. The electronic and magnetic changes depend on the concentration of hydrogen atoms as well as on hydrogenation rate [15]. In advance, the effects of hydrogenation had been studied for graphene, silicene, and germane [16,17,18].
As tin is a tetravalent element in group IV of the periodic table, it is plausible to functionalize it by adding a donor impurity from group V such as Phosphorus and an acceptor impurity from group II such as Aluminum. It has been reported that Al adatom dan P adatom adsorption on graphene, germanene, and silicene change their electronic structure and magnetic properties [19,20].
Controlling the electronic properties of stanene has encouraged enormous interest, which can lead to design and fabricate high performance stanene based electronic devices. However, not all potential doped stanene systems have been explored. Therefore, in this work, we present first-principles DFT calculations that compare the magnetic properties of flat stanene with different states: pristine, hydrogenated, Al-doped, P-doped, hydrogenated Al-doped, and hydrogenated P-doped. Hopefully, this work can trigger the synthesis of these stanene systems.
2. Methods
All the calculations were performed within the framework of spin-polarized plane-wave Density Functional Theory (DFT) implemented in the ABINIT package [21]. We used Generalized Gradient approximation (GGA) for the plane-wave basis set and Hamann-type pseudopotentials with Perdew–Burke–Ernzerhof parametrization [22]. An cutoff energy of 500 eV was used for the plane-wave expansion of the electronic wave function. The k-point was set to 25 × 25 × 1 Monkhorst-Pack mesh for the Brillouin zone integration [23]. Stanene was hydrogenated by adding hydrogen atoms to each Sn atom with a zig-zag position. Initially, We considered three high symmetry adsorption sites of Al adatom and P adatom on Stanane. The hollow site (H-site) above the center of hexagonal Tin (Sn) rings, the Bridge site (B-site) at the midpoint of the Sn-Sn bond, and the Top site (T-site) directly above one Sn atom. Then, we calculated the formation energy of Al/P–doped stanene with the three possible sites to obtain the most stable one. Our results show that the lowest one is stanene with Al/P with a hollow site which indicating hollow is the most favorable site for stanene with these transition metal adatoms. It was reported that hollow site is the most stable position of other transition metal adatoms (V, Cr, Mn, Fe, Co, Ni) adsorbed on stanene [17]. Therefore, we only calculated the electronic structure and magnetic properties of pristine stanene and hydrogenated stanene with Al/P adatom at the hollow site. We compared the electronic structure and magnetic properties of six stanene systems, i.e: Pristine stanene, hydrogenated stanene, Al-doped stanene, P-doped stanene, Al-doped hydrogenated stanene, and P-doped hydrogenated stanene.
3. Results and Discussion
To know which magnetic state is more favorable, we calculated the energy of nonmagnetic, ferromagnetic, and antiferromagnetic state of all our stanene systems. The results show that energy of antiferromagnetic state for all stanene system is higher than that of nonmagnetic and ferromagnetic states, which means there is no antiferromagnetic in these stanene systems. Thus, in Table 1, we only show energy difference ∆E between the ferromagnetic (FM) and nonmagentic (NM) which can be expressed as
where E(NM) is the total energy of a nonmagnetic state, E(FM) is the total energy of the ferromagnetic state. A negative value of ∆E implies an NM state is preferred, while a positive value of ∆E implies an FM state is preferred. From Table 1, it is obvious that only pristine stanene is favor nonmagnetic state while others are in favor of ferromagnetic.

Table 1.
Calculated energy difference between feromagnetic state and nonmagnetic state of stanene systems.
To know the magnetic strength of each stanene system, we calculated their magnetic moment. Figure 1 denotes hierarchy of the magnetic moment of the stanene systems from small to large. Hydrogenation enhances the magnetic moment most significantly. Effect of P atom on the magnetic moment of stanene slightly less than that of H atom but larger than that of Al atom.
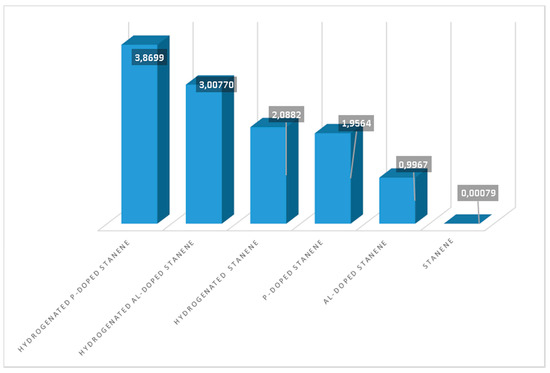
Figure 1.
Magnetic moment of the six stanene systems: pure stanene, Al-doped stanene, P-doped stanene, hydrogenated stanene, hydrogenated Al-doped stanene, and hydrogenated P-doped stanene.
To illustrate the corresponding electronic properties, the spin-resolved density of states of the stanene systems are given in Figure 2, Figure 3, Figure 4, Figure 5 and Figure 6. The Density of State of pure stanene (Figure 2) shows Dirac cone at Fermi energy. Sn 5s orbital dominate valence band and 5p orbitals have the major contribution in the conduction band as reported by previous works [24]. We note that the density of states are nearly the same for up and down spins at the valence band, there are only very small asymmetries at some part of the conduction band. This consistent with its negligible magnetic moment (Table 1).
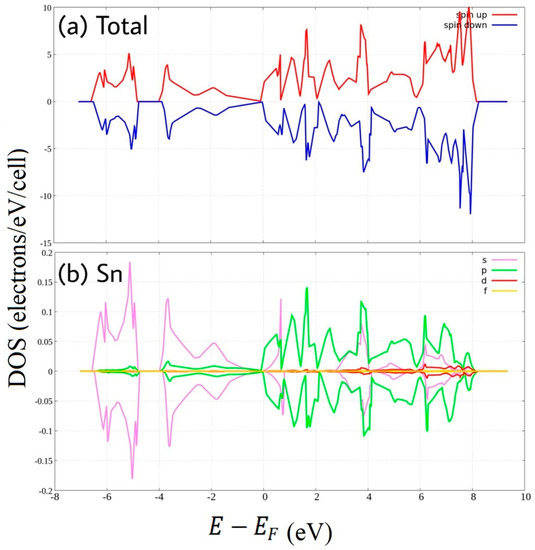
Figure 2.
Spin-polarized Density of State of pure stanene. (a) total DOS (b) PDOS of Sn atom.
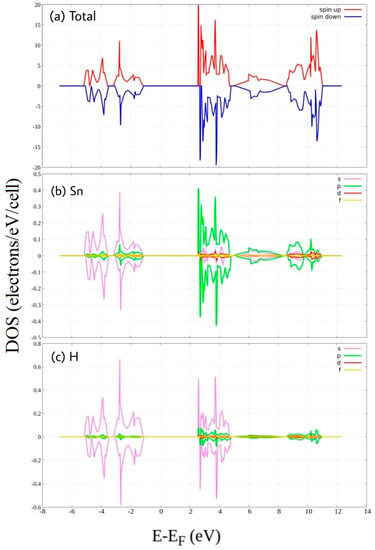
Figure 3.
Spin-polarized Density of State of hydrogenated stanene. (a) total DOS, and PDOS of (b) Sn atom and (c) H atom.
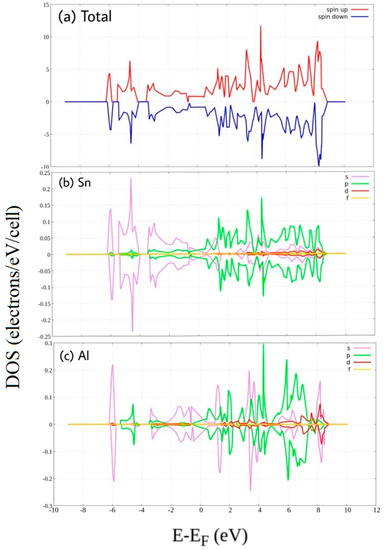
Figure 4.
Spin-polarized Density of State of Al-doped stanene. (a) total DOS, and PDOS of (b) Sn atom and (c) P atom.
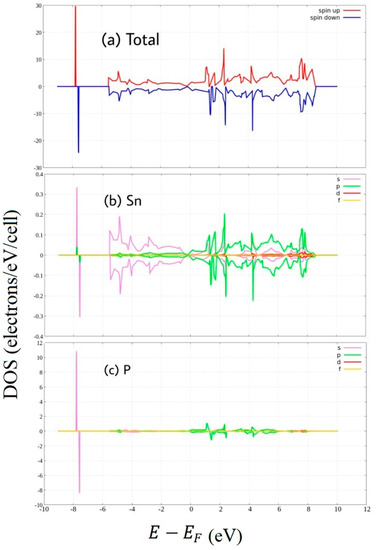
Figure 5.
Spin-polarized Density of State of P-doped stanene. (a) total DOS, and PDOS of (b) Sn atom and (c) P atom.
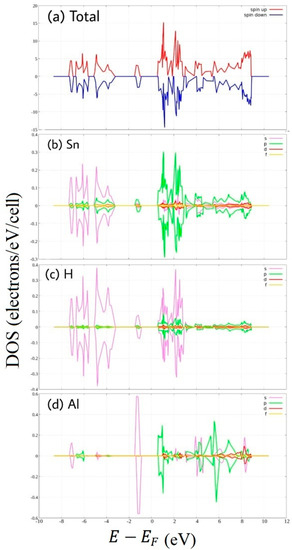
Figure 6.
Spin-polarized Density of State of hydrogenated Al-doped stanene. (a) total DOS, and PDOS of (b) Sn, (c) H atom, and (d) Al atom.
Hydrogenation clearly creates a wide gap around the Fermi energy of stanene (Figure 3). Above fermi energy, peak belongs to H 1s states and Sn 5p states overlap in the same energy region or change in the same way indicating hybridization. Also below fermi energy, H 1s states hybridize with Sn 5s states. This hybridization is attributed to the covalent bonding of Sn-H, as reported by previous first principle calculations [25]. Formation of energy gap due to hydrogenation has been reported previously in other 2D honeycomb materials such as graphene [26,27], germanene, and silicene [28]. It is also clearly seen that the up and down DOS have been “shifted” with respect to each other in the region from 2.5 eV to 5 eV which arise primarily from the Sn 5p states and H 1s states. This shift is responsible for the increase of the magnetic moment of stanene.
From PDOS shown in Figure 4, we can see that orbital 3s and orbital 3p of Al atoms contribute dominantly alternately at both the valence band and the conduction band. Al doping does not induce band gap but eliminates the Dirac cone at Fermi energy. Near to Fermi energy, Al 3s across Fermi energy to upper energy, on the other hand, Al 3p across to lower energy. This orbital overlap leads to a metallic behavior. This consistent with previous work [29]. A similar case was also reported for Al-doped graphene [20]. We also can see up-down asymmetries resulting from unpaired 3p electrons, at some part of the conduction band leads to an increase of the magnetic moment of Al doped stanene. Hence, the valance band of Al-doped stanene mainly consists of Sn 5s states combined with Al 3s state and Al 3p state, while its conduction band is composed of Sn 5p states combined also with Al 3s state and Al 3p state.
Figure 5 shows the density of state of P-doped stanene. Unlike Al-doped stanene, P-doped stanene does not clearly exhibit metallic behavior. In the vicinity of the Fermi level, the shape of the density of state similar to that of pure stanene where Dirac cone is still present. At low energy, the P atom induces a new state. From this figure, one can see asymmetric PDOS of the P atom which mainly consists of P 3p states. The asymmetry arising from unpaired 3p electrons of phosphorus results in the magnetic moment enhancement of stanene.
In Al-doped hydrogenated stanene, we can see new middle states appear as denoted by Figure 6. The midgap electronic states are due mainly to Al 3s states, slightly to H 1s states and to Sn 3p states. The valence band is composed predominantly of Sn 3s states and H 1s states. While, the conduction band is formed from Sn 3p states, H 1s, and Al 3p states.
Impurity state also appears in P-doped hydrogenated stanene (Figure 7). The impurity states, which are close to the conduction band, due to extra electrons make the stanene become n-type semiconductor. P atoms as pentavalent impurities give free electrons. The unpaired free electrons produce spin-asymmetry of the Density of States. We can see the obvious asymmetry of the PDOS of the P atom at the conduction band which mainly consists of P 3p states. This is the reason for P-doped hydrogenated stanene becomes the most magnetic stanene system among these six stanene systems as denoted by Figure 1.
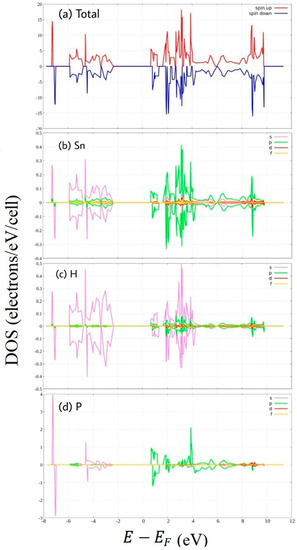
Figure 7.
Spin-polarized Density of State of hydrogenated P-doped stanene. (a) total DOS, and PDOS of (b) Sn atom, (c) H atom, and (d) P atom.
Figure 8 displays the charge density of six stanene systems. As shown in Figure 8a electron density concentrated in the middle between two adjacent tin atoms which indicating covalent bonding [1]. Hydrogen atoms change the distribution of charge density (Figure 8b), higher electron density (yellow color) is concentrated between two farther Sn atoms (next nearest neighbor) which is seemingly making sigma bond, while bonding between two adjacent electrons are broken resulting the electrons localized and unpaired which leads to bandgap opening (Figure 3) and magnetic moment enhancement (Figure 1). These results agree with previous first principle calculations [15]. Nonmagnetic metal to magnetic semiconducting transition due to adsorbed hydrogen atoms also occur in graphene [30] and silicene [31]. The interstitial Al atom (Figure 8c) and the P atom (Figure 8d) do not make bonding with the Sn atom. The negative charge is not seen in the charge density of Al-doped stanene indicating Al atom provides hole. In contrast, it is seen that the P atom donates an electron to the P-doped stanene. The electrons are localized around the phosphorous atom. This finding is consistent with previous DFT study [29]. Figure 8e shows the charge density of Al-doped hydrogenated stanene. It is seen that the Al atom at the hollow site separates the electron density of the opposite Sn atoms and prevents them from connecting. This induces an impurity state at the bandgap of Al-doped hydrogenated stanene (see Figure 6) and creates unpaired electrons. In P-doped hydrogenated stanene, density of electrons inhabit p orbital around P atom increases and the electrons more localized than that in P-doped stanene without hydrogen passivation (see Figure 8f). P atom at the hollow site also separates the electron density of the adjacent Sn atoms and hinders them to connect each other which in turn produces unpaired electrons and responsible for magnetic moment enhancement.
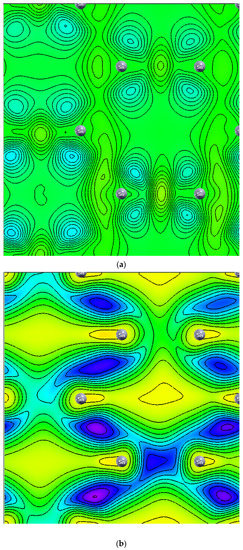
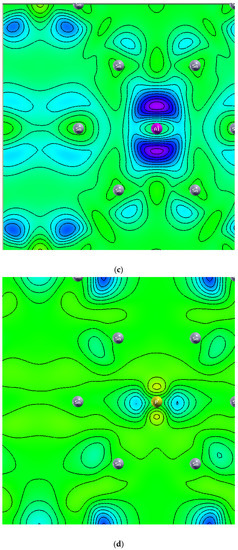
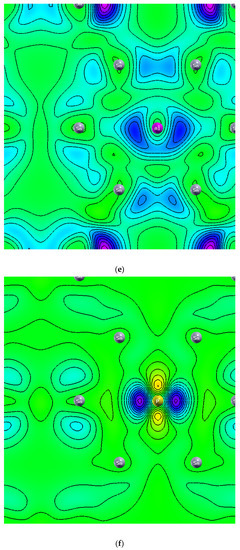
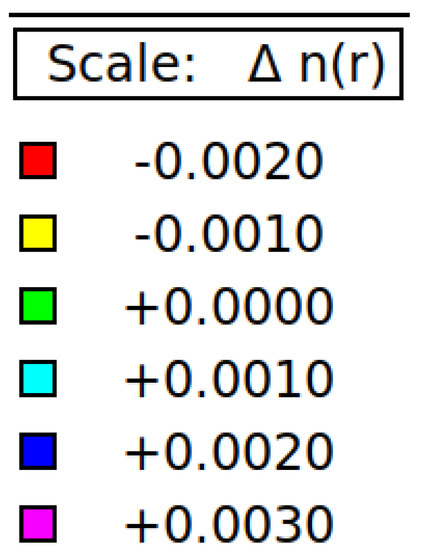
Figure 8.
Charge density of: (a) Pure stanene, (b) Hydrogenated stanene, (c) Al-doped stanene, (d) P-doped stanene, (e) Al-doped hydrogenated stanene, and (f) P-doped hydrogenated stanene.
4. Conclusions
We have discussed the roles of hydrogen atoms and Al/P doping on nonmagnetic metal to magnetic semiconducting transition of the stanene systems. Hydrogen passivation induces a wide bandgap and increases magnetic moment that involved H 1s states and Sn 5p states hybridization. Aluminum atom and Phosphorous atom at hollow site act as a barrier against the connection of neighboring tin atoms resulting magnetic moment enhancement of the stanene systems. By the combination of hydrogenation and doping of Al/P atom, a stanene-based magnetic semiconductor can be obtained.
Author Contributions
Conceptualization, M.A.P.; methodology, M.A.P., V.K.R.S.; software, V.K.R.S., I., S.A.P.; validation, V.K.R.S., I. and S.A.P.; formal analysis, M.A.P., V.K.R.S., I.; investigation, M.A.P., V.K.R.S., I.; resources, V.K.R.S.; data curation, M.A.P. and I.; writing—original draft preparation, V.K.R.S. and M.A.P.; writing—review and editing, M.A.P., A. and M.N.; visualization, V.K.R.S. and I.; supervision, M.A.P.; project administration, M.A.P.; funding acquisition, A., M.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Education and Culture of Indonesia through reasearch grant with contract number 167/SP2H/LT/DRPM/2019.
Data Availability Statement
Data available in a publicly accessible repository.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lyu, J.; Zhang, S.; Zhang, C.; Wang, P. Stanene: A Promising Material for New Electronic and Spintronic Applications. Ann. Phys. 2019, 531, 1900017. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, B.; Zhang, H.; Wang, J.; Xu, G.; Tang, P.; Duan, W.; Zhang, S. Large-Gap Quantum Spin Hall Insulators in Tin Films. Phys. Rev. Lett. 2013, 111, 136804. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Zhang, H.; Shao, H.; Xu, Y.; Zhang, X.; Zhu, H. Low lattice thermal conductivity of stanene. Sci. Rep. 2016, 6, 20225. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Y.; Zhang, S. Two-dimensional time-reversal-invariant topological superconductivity in a doped quantum spin-Hall insulator. Phys. Rev. B Condens. Matter Mater. Phys. 2014, 90, 054503. [Google Scholar] [CrossRef]
- Rachel, S.; Ezawa, M. Giant magnetoresistance and perfect spin filter in silicene, germanene, and stanene. Phys. Rev. B 2014, 89, 195303. [Google Scholar] [CrossRef]
- Molle, A.; Goldberger, J.; Houssa, M.; Xu, Y.; Zhang, S. Buckled two-dimensional Xene sheets. Nat. Mater. 2017, 16, 163–169. [Google Scholar] [CrossRef]
- Deng, J.; Xia, B.; Ma, X.; Chen, H.; Shan, H.; Zhai, X.; Li, B.; Zhao, A.; Xu, Y.; Duan, W.; et al. Epitaxial growth of ultraflat stanene with topological band inversion. Nat. Mater. 2018, 17, 1081. [Google Scholar] [CrossRef]
- Ahmed, R.; Nakagawa, T.; Mizuno, S. Structure determination of ultra-flat stanene on Cu(111) using low energy electron diffraction. Surf. Sci. 2020, 691, 121498. [Google Scholar] [CrossRef]
- Yuhara, J.; Fujii, Y.; Nishino, K.; Isobe, N.; Nakatake, M.; Xian, L.; Rubio, A.; Lay, G. Le Large area planar stanene epitaxially grown on Ag (1 1 1). 2D Mater. 2018, 5, 025002. [Google Scholar] [CrossRef]
- Modarresi, M.; Kakoee, A.; Mogulkoc, Y.; Roknabadi, M.R. Effect of external strain on electronic structure of stanene. Comput. Mater. Sci. 2015, 101, 164–167. [Google Scholar] [CrossRef]
- Wang, D.; Chen, L.; Shi, C.; Grazianetti, C.; Cinquanta, E.; Molle, A.; Ren, C.; Ji, W.; Zhang, C.; Li, P.; et al. The effects of biaxial strain and electric fi eld on the electronic properties in stanene. Mater. Res. Express 2016, 3, 105008. [Google Scholar]
- Gou, J.; Kong, L.; Li, H.; Zhong, Q.; Li, W.; Cheng, P.; Chen, L. Strain-Induced Band Engineering in Monolayer Stanene on Sb(111). Phys. Rev. Mater. 2017, 1, 054004. [Google Scholar] [CrossRef]
- Van Den Broek, B.; Houssa, M.; Scalise, E.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. Two-dimensional hexagonal tin: Ab initio geometry, stability, electronic structure and functionalization. 2D Mater. 2014, 1, 021004. [Google Scholar] [CrossRef]
- Naqvi, S.R.; Hussain, T.; Luo, W.; Ahuja, R. Exploring Doping Characteristics of Various Adatoms on Single-layer Stanene. J. Phys. Chem. C 2017, 121, 7667. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C. Tunable electronic structures and magnetic properties in two-dimensional stanene with hydrogenation. Mater. Chem. Phys. 2016, 173, 246. [Google Scholar] [CrossRef]
- Drissi, L.B.; Saidi, E.H.; Bousmina, M.; Fassi-Fehri, O. DFT investigations of the hydrogenation effect on silicene/graphene hybrids. J. Phys. Condens. Matter 2012, 24, 485502. [Google Scholar] [CrossRef] [PubMed]
- Houssa, M.; Scalise, E.; Sankaran, K.; Pourtois, G.; Afanas’Ev, V.V.; Stesmans, A. Electronic properties of hydrogenated silicene and germanene. Appl. Phys. Lett. 2011, 98, 2011–2014. [Google Scholar] [CrossRef]
- Boukhvalov, D.W.; Katsnelson, M.I.; Lichtenstein, A.I. Hydrogen on graphene: Electronic structure, total energy, structural distortions and magnetism from first-principles calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 1–7. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, D.; Xie, F.; Zheng, X.; Wang, H.; Long, M. First-principles study on the magnetic and electronic properties of Al or P doped armchair silicene nanoribbons. Phys. Lett. A 2017, 381, 2097–2102. [Google Scholar] [CrossRef]
- Romero, M.T.; Alvarado, Y.A.; Garcia, C.R. First Principles Calculations of Graphene Doped with Al, P and Si Heteroatoms. Nano Hybrids Compos. 2017, 16, 52–55. [Google Scholar] [CrossRef]
- Gonze, X.; Amadon, B.; Anglade, P.M.; Beuken, J.M.; Bottin, F.; Boulanger, P.; Bruneval, F.; Caliste, D.; Caracas, R.; Côté, M.; et al. ABINIT: First-principles approach to material and nanosystem properties. Comput. Phys. Commun. 2009, 180, 2582–2615. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Pack, J.D.; Monkhorst, H.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Garg, P.; Choudhuri, I.; Mahata, A.; Pathak, B. Band Gap Opening in Stanene Induced by Patterned B-N Doping. Phys. Chem. Chem. Phys. 2017, 19, 3660. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Long, L.; Xu, C.; Yuan, J. First-principles study on the structure and electronic properties of stanane under electric fields. Mater. Res. Express 2018, 5, 065023. [Google Scholar] [CrossRef]
- Gao, H.; Wang, L.; Zhao, J.; Ding, F.; Lu, J. Band Gap Tuning of Hydrogenated Graphene: H Coverage and Configuration Dependence. J. Phys. Chem. C 2011, 115, 3236–3242. [Google Scholar] [CrossRef]
- Son, J.; Lee, S.; Kim, S.J.; Park, B.C.; Lee, H.; Kim, S.; Kim, J.H. Hydrogenated monolayer graphene with reversible and tunable wide band gap and its field-effect transistor. Nat. Commun. 2016, 7, 13261. [Google Scholar] [CrossRef]
- Trivedi, S.; Srivastava, A.; Kurchania, R. Silicene and germanene: A first principle study of electronic structure and effect of hydrogenation-passivation. J. Comput. Theor. Nanosci. 2014, 11, 781–788. [Google Scholar] [CrossRef]
- Abbasi, A. DFT study of the effects of Al-P pair doping on the structural and electronic properties of stanene nanosheets. Phys. E Low Dimens. Syst. Nanostruct. 2019, 108, 34. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Q.; Sun, Q.; Chen, X.S.; Kawazoe, Y.; Jena, P. Ferromagnetism in Semihydrogenated Graphene Sheet. Nano Lett. 2009, 9, 3867–3870. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, C. The electronic and magnetic properties of functionalized silicene: A first-principles study. Nanoscale Res. Lett. 2012, 7, 422. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).