
Molecular Dynamics Simulation on the Interaction between Palygorskite Coating and Linear Chain Alkane Base Lubricant


Abstract
:
1. Introduction
2. Models and Simulation Methods
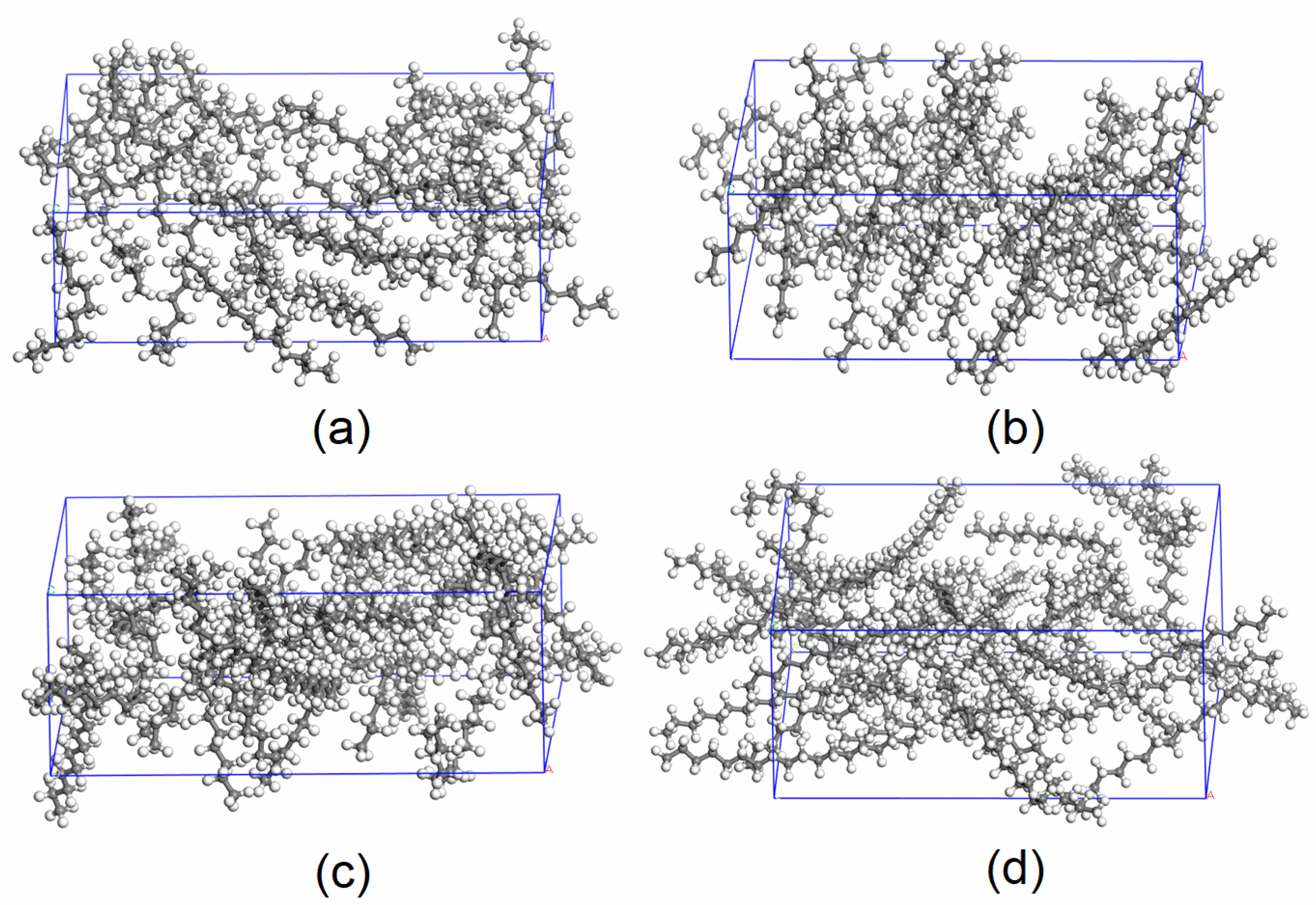
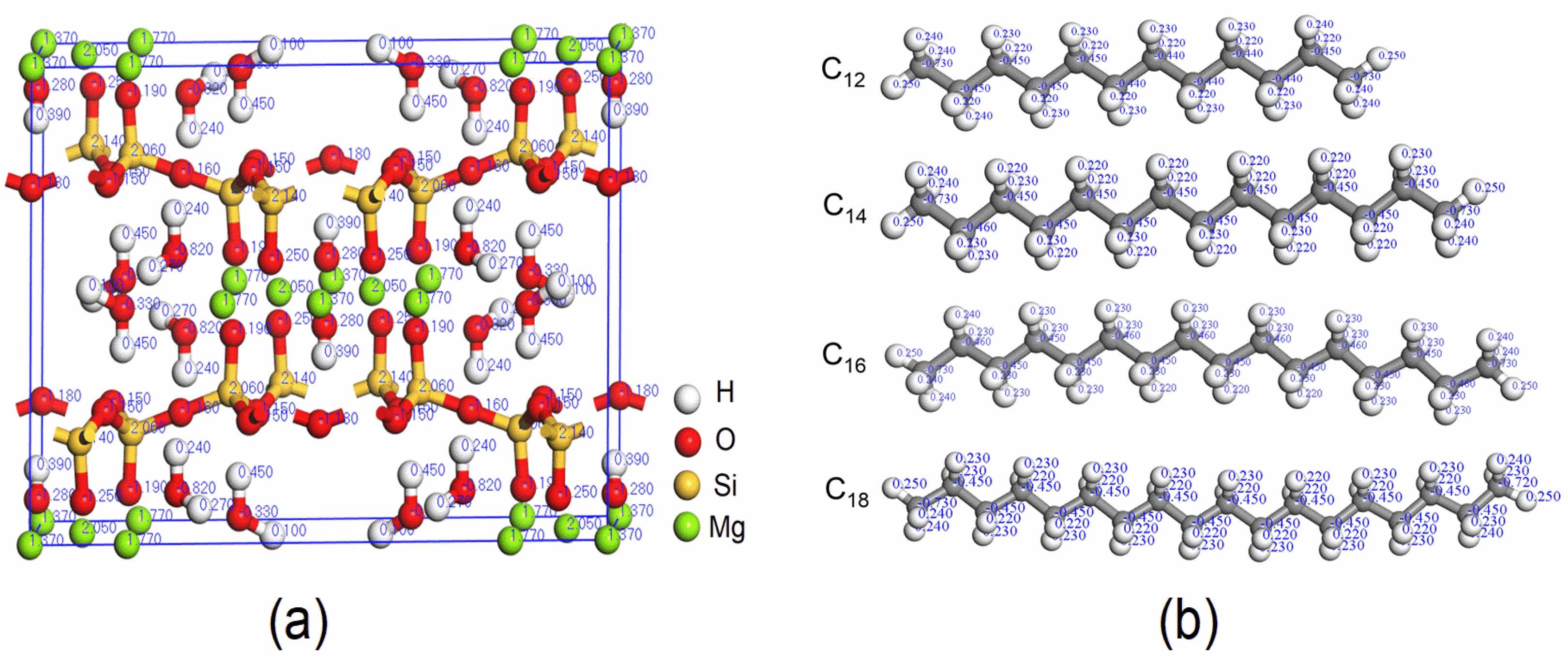
2.1. Model Construction
2.2. Simulation Methods
3. Results and Discussion
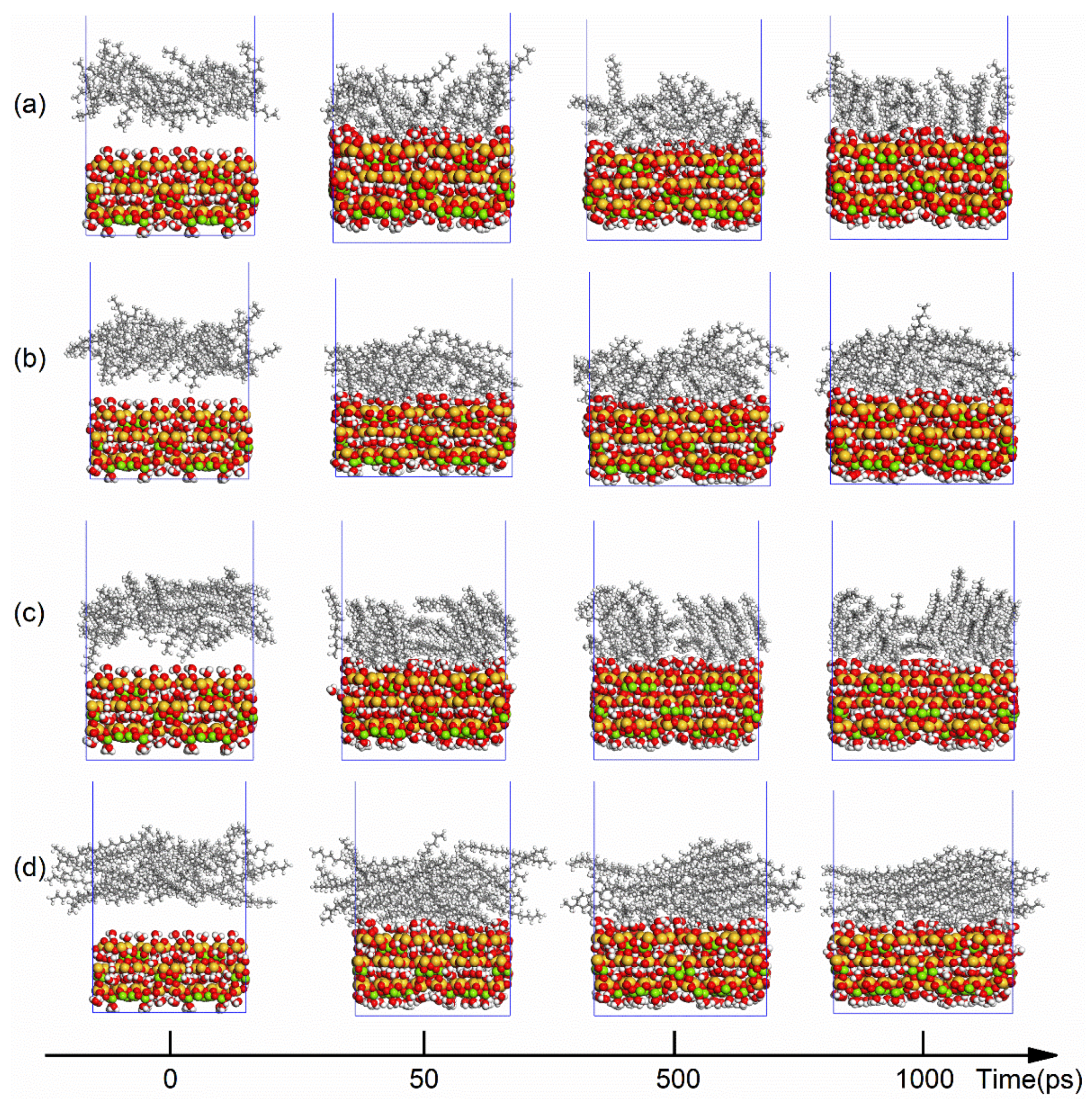
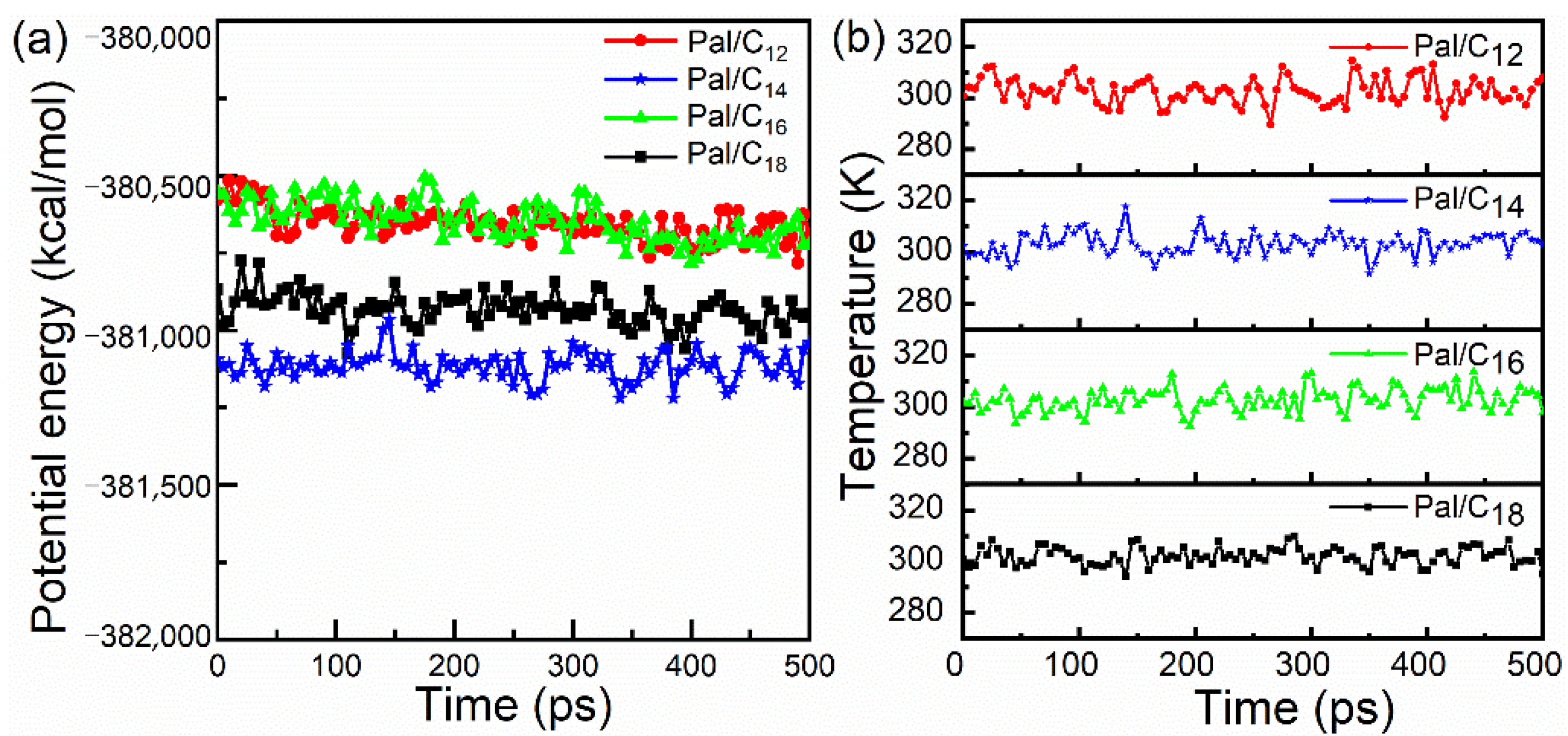
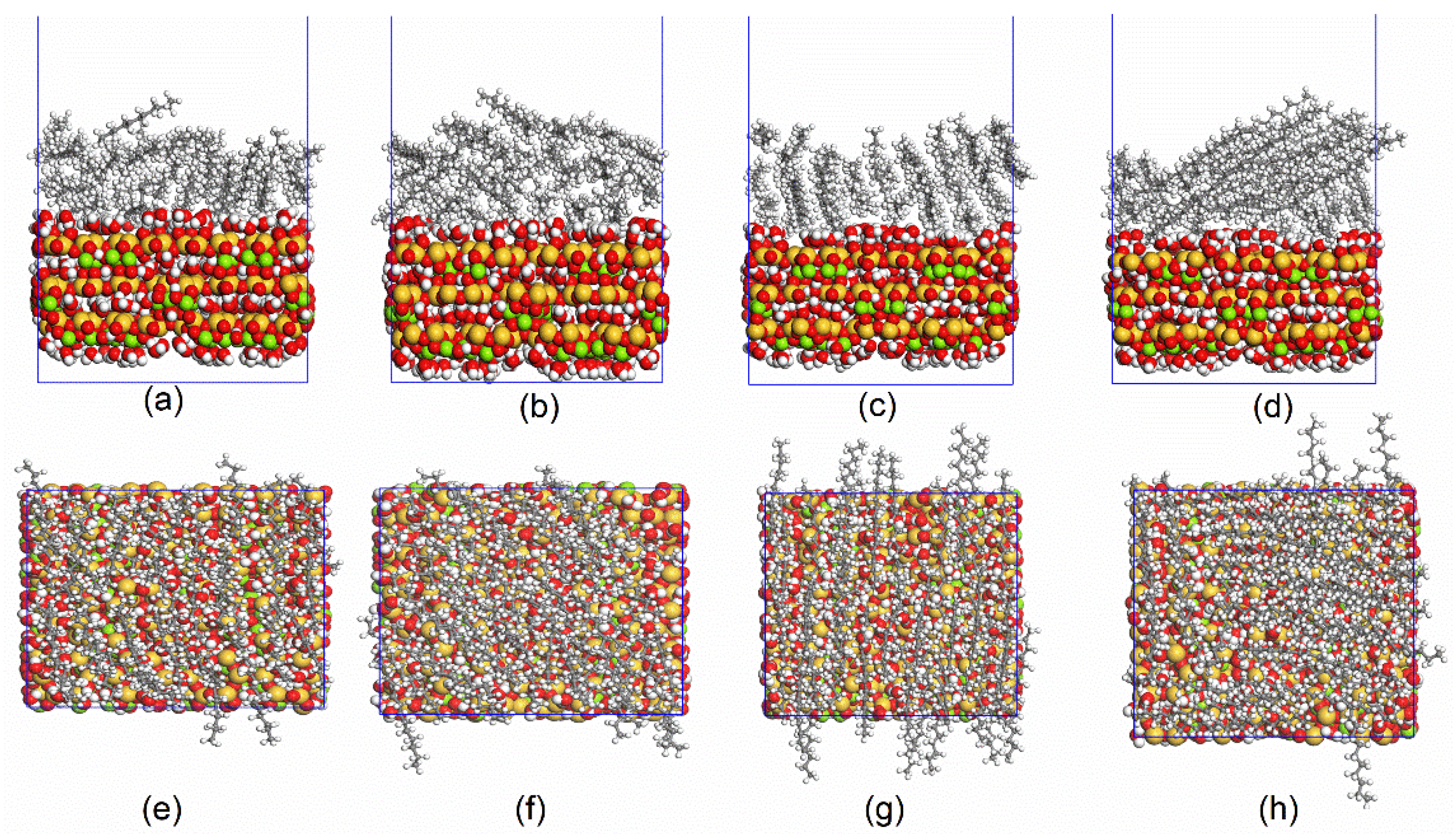
3.1. MD Simulation and System Equilibrium Process
3.2. Interaction Energies
3.3. Mean Square Displacement and Diffusion Coefficient
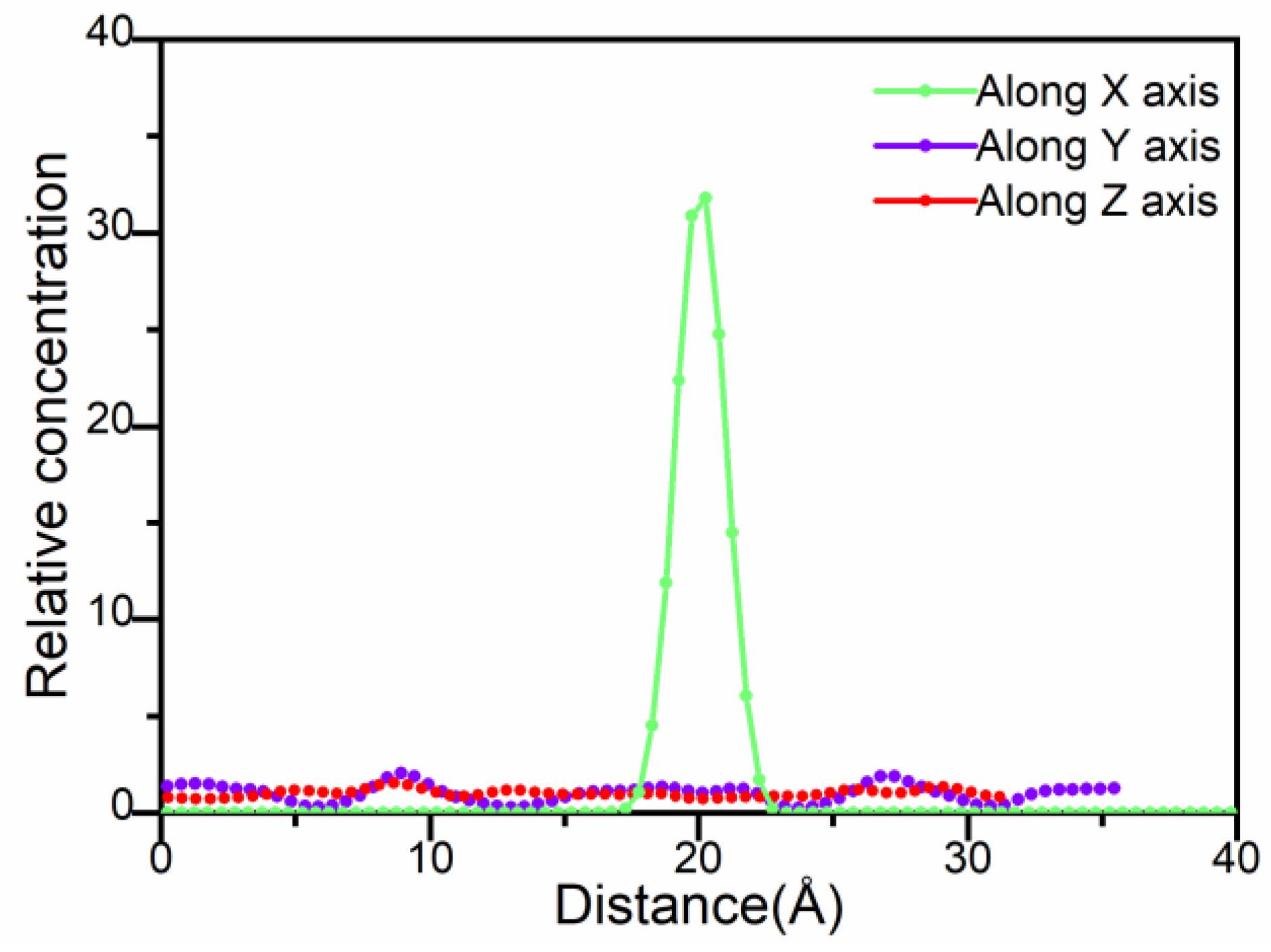
3.4. Water Molecules in Palygorskite
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yang, L.; Li, Y.; Cai, J.; Ding, X.; Cao, Y. The formation mechanisms for auto-restoration tribofilm at the presence of palygorskite-Ag nanoparticles. Integr. Ferroelectr. 2017, 182, 110–118. [Google Scholar] [CrossRef]
- Gaines, R.V.; Skinner, H.C.W.; Foord, E.E.; Mason, B.; Rosenzweig, A. Dana’s New Mineralogy, 8th ed.; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Galan, E. Properties and applications of palygorskite-sepiolite clays. Clay Miner. 1996, 31, 443–453. [Google Scholar] [CrossRef]
- Galán, E. A New Approach to Compositional Limits for Sepiolite and Palygorskite. Clays Clay Miner. 1999, 47, 399–409. [Google Scholar] [CrossRef]
- Pan, Z.; Zhao, A.; Pan, R. The Crystallography and Mineralogy; Geological Press: Beijing, China, 1998. [Google Scholar]
- Middea, A.; Fernandes, T.L.; Neumann, R.; Gomes, O.D.F.; Spinelli, L.S. Evaluation of Fe(III) adsorption onto palygorskite surfaces. Appl. Surf. Sci. 2013, 282, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Zhang, P.; Wang, Y.; Wen, K.; Su, X.; Zhu, R.; He, H.; Xi, Y. Effect of acid activation of palygorskite on their toluene adsorption behaviors. Appl. Clay Sci. 2018, 159, 60–67. [Google Scholar] [CrossRef]
- Sarkar, B.; Megharaj, M.; Xi, Y.; Naidu, R. Surface charge characteristics of organo-palygorskites and adsorption of p-nitrophenol in flow-through reactor system. Chem. Eng. J. 2012, 185–186, 35–43. [Google Scholar] [CrossRef]
- Silva, A.F.; Burggraeve, A.; Denon, Q.; Van Der Meeren, P.; Sandler, N.; Kerkhof, T.V.D.; Hellings, M.; Vervaet, C.; Remon, J.P.; Lopes, J.A.; et al. Particle sizing measurements in pharmaceutical applications: Comparison of in-process methods versus off-line methods. Eur. J. Pharm. Biopharm. 2013, 85, 1006–1018. [Google Scholar] [CrossRef]
- Chen, M.; Jiang, W.; Wang, F.; Shen, P.; Ma, P.; Gu, J.; Mao, J.; Li, F. Synthesis of highly hydrophobic floating magnetic polymer nanocomposites for the removal of oils from water surface. Appl. Surf. Sci. 2013, 286, 249–256. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Z.; Yao, C.; Lu, X.; Zhao, X.; Ni, C. Attapulgite-CeO2/MoS2 ternary nanocomposite for photocatalytic oxidative desulfurization. Appl. Surf. Sci. 2016, 364, 589–596. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Wang, A. Synthesis and characterization of chitosan-g-poly(acrylic acid)/attapulgite superabsorbent composites. Carbohydr. Polym. 2007, 68, 367–374. [Google Scholar] [CrossRef]
- Pei, M.; Pan, C.; Wu, D.; Liu, P. Surface hydrophilic-hydrophobic reversal coatings of polydimethylsiloxane-palygorskite nanosponges. Appl. Clay Sci. 2020, 189, 105546. [Google Scholar] [CrossRef]
- Zhang, P.; Tian, N.; Zhang, J.; Wang, A. Effects of modification of palygorskite on superamphiphobicity and microstructure of palygorskite@fluorinated polysiloxane superamphiphobic coatings. Appl. Clay Sci. 2018, 160, 144–152. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J. Durable and self-healing superamphiphobic coatings repellent even to hot liquids. Chem. Commun. 2016, 52, 2744–2747. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Du, M.; Zheng, Q. Dopamine/Silica Nanoparticle Assembled, Microscale Porous Structure for Versatile Superamphiphobic Coating. ACS Nano 2016, 10, 2910–2921. [Google Scholar] [CrossRef]
- Dong, J.; Wang, Q.; Zhang, Y.; Zhu, Z.; Xu, X.; Zhang, J.; Wang, A. Colorful Superamphiphobic Coatings with Low Sliding Angles and High Durability Based on Natural Nanorods. ACS Appl. Mater. Interfaces 2017, 9, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Dong, S.; Li, B.; Wei, X.; Zhang, J. Durable and fluorine-free superhydrophobic coatings from palygorskite-rich spent bleaching earth. Appl. Clay Sci. 2018, 157, 237–247. [Google Scholar] [CrossRef]
- Dong, J.; Zhu, Q.; Wei, Q.; Zheng, B.; Li, S.; Zhang, J. A comparative study about superamphiphobicity and stability of superamphiphobic coatings based on Palygorskite. Appl. Clay Sci. 2018, 165, 8–16. [Google Scholar] [CrossRef]
- Li, X.-J.; Yan, C.-J.; Luo, W.-J.; Gao, Q.; Zhou, Q.; Liu, C.; Zhou, S. Exceptional cerium(III) adsorption performance of poly(acrylic acid) brushes-decorated attapulgite with abundant and highly accessible binding sites. Chem. Eng. J. 2016, 284, 333–342. [Google Scholar] [CrossRef]
- Lakbita, O.; Rhouta, B.; Maury, F.; Senocq, F.; Amjoud, M.; Daoudi, L. Influence of the crystal structure of Ag2CO3 on the photocatalytic activity under visible light of Ag2CO3-Palygorskite nanocomposite material. Appl. Surf. Sci. 2019, 464, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, A. Electrokinetic and Colloidal Properties of Homogenized and Unhomogenized Palygorskite in the Presence of Electrolytes. J. Chem. Eng. Data 2012, 57, 1586–1593. [Google Scholar] [CrossRef]
- Huang, D.; Wang, W.; Xu, J.; Wang, A. Mechanical and water resistance properties of chitosan/poly(vinyl alcohol) films reinforced with attapulgite dispersed by high-pressure homogenization. Chem. Eng. J. 2012, 210, 166–172. [Google Scholar] [CrossRef]
- Lai, S.-Q.; Li, T.-S.; Liu, X.-J.; Lv, R.-G. A Study on the Friction and Wear Behavior of PTFE Filled with Acid Treated Nano-Attapulgite. Macromol. Mater. Eng. 2004, 289, 916–922. [Google Scholar] [CrossRef]
- Wang, K.; Wu, H.; Wang, H.; Liu, Y.; Yang, L.; Zhao, L. Tribological properties of novel palygorskite nanoplatelets used as oil-based lubricant additives. Friction 2021, 9, 332–343. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, Y.; Li, Y.; Cai, J.; Cao, Y. Effect of Ag Content in Palygorskite/Ag Blending Nanocomposite on Friction and Wear Properties of 45 Mild Steel Tribopair. Integr. Ferroelectr. 2013, 145, 10–16. [Google Scholar] [CrossRef]
- Fois, E.; Gamba, A.; Tilocca, A. On the unusual stability of Maya blue paint: Molecular dynamics simulations. Microporous Mesoporous Mater. 2003, 57, 263–272. [Google Scholar] [CrossRef]
- Chiari, G.; Giustetto, R.; Ricchiardi, G. Crystal structure refinements of palygorskite and Maya Blue from molecular modelling and powder synchrotron diffraction. Eur. J. Miner. 2003, 15, 21–33. [Google Scholar] [CrossRef]
- Giustetto, R.; Vitillo, J.G.; Corazzari, I.; Turci, F. Evolution and Reversibility of Host/Guest Interactions with Temperature Changes in a Methyl Red@Palygorskite Polyfunctional Hybrid Nanocomposite. J. Phys. Chem. C 2014, 118, 19322–19337. [Google Scholar] [CrossRef]
- Bu, J.; Teresa, R.G.; Brown, K.; Sanchez, F. Adsorption mechanisms of cesium at calcium-silicate-hydrate surfaces using molecular dynamics simulations. J. Nucl. Mater. 2019, 515, 35–51. [Google Scholar] [CrossRef]
- Zheng, D.; Wang, X.; Zhang, M.; Liu, Z.; Ju, C. Anticorrosion and lubricating properties of a fully green lubricant. Tribol. Int. 2019, 130, 324–333. [Google Scholar] [CrossRef]
- Accelrys Inc. Materials Studio Version 8.0; Accelrys Inc.: San Diego, CA, USA, 2014; Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio/ (accessed on 27 February 2021).
- Post, J.E.; Heaney, P.J. Synchrotron powder X-ray diffraction study of the structure and dehydration behavior of palygorskite. Am. Miner. 2008, 93, 667–675. [Google Scholar] [CrossRef]
- Rajan, H.; Uchida, H.; Bryan, D.L.; Swaminathan, R.; Downs, R.T.; Hall-Wallace, M. The American mineralogist crystal structure database. Am. Miner. 2003, 88, 247–250. [Google Scholar]
- Berro, H.; Fillot, N.; Vergne, P. Molecular dynamics simulation of surface energy and ZDDP effects on friction in nano-scale lubricated contacts. Tribol. Int. 2010, 43, 1811–1822. [Google Scholar] [CrossRef]
- Jaishankar, A.; Jusufi, A.; Vreeland, J.L.; Deighton, S.P.; Pellettiere, J.R.; Schilowitz, A.M. Adsorption of Stearic Acid at the Iron Oxide/Oil Interface: Theory, Experiments, and Modeling. Langmuir 2019, 35, 2033–2046. [Google Scholar] [CrossRef]
- Toth, R.; Coslanich, A.; Ferrone, M.; Fermeglia, M.; Pricl, S.; Miertus, S.; Chiellini, E. Computer simulation of polypropylene/organoclay nanocomposites: Characterization of atomic scale structure and prediction of binding energy. Polymer 2004, 45, 8075–8083. [Google Scholar] [CrossRef]
- Castonguay, L.A.; Rappe, A.K. Ziegler-Natta catalysis. A theoretical study of the isotactic polymerization of propylene. J. Am. Chem. Soc. 1992, 114, 5832–5842. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Zhang, S.-G.; Wang, F.-Y.; Tan, X.-Y. Molecular dynamics simulation the hydroxyapatite scale inhibition mechanism of water-soluble polymers. J. Theor. Comput. Chem. 2010, 9, 889–902. [Google Scholar] [CrossRef]
- Zeng, J.-P.; Qian, X.-R.; Wang, F.-H.; Shao, J.-L.; Bai, Y.-S. Molecular dynamics simulation on the interaction mechanism between polymer inhibitors and calcium phosphate. J. Chem. Sci. 2014, 126, 649–658. [Google Scholar] [CrossRef]
- Nymand, T.M.; Linse, P. Ewald summation and reaction field methods for potentials with atomic charges, dipoles, and polarizabilities. J. Chem. Phys. 2000, 112, 6152–6160. [Google Scholar] [CrossRef]
- Bhowmik, R.; Katti, K.S.; Katti, D. Molecular dynamics simulation of hydroxyapatite–polyacrylic acid interfaces. Polymer 2007, 48, 664–674. [Google Scholar] [CrossRef]
- Tong, Z.; Xie, Y.; Zhang, Y. Molecular dynamics simulation on the interaction between polymer inhibitors and β-dicalcium silicate surface. J. Mol. Liq. 2018, 259, 65–75. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, J.; Gong, X. Molecular dynamics simulation of interaction between benzotriazoles and cuprous oxide crystal. Comput. Theor. Chem. 2011, 963, 110–114. [Google Scholar] [CrossRef]
- Zeng, J.-P.; Wang, F.-H.; Zhou, C.; Gong, X.-D. Molecular Dynamics Simulation on Scale Inhibition Mechanism of Polyepoxysuccinic Acid to Calcium Sulphate. Chin. J. Chem. Phys. 2012, 25, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Keer, L.M.; Lu, J.; Song, B.; Gu, L. Molecular dynamics simulations of the rheological properties of graphene–PAO nanofluids. J. Mater. Sci. 2018, 53, 15969–15976. [Google Scholar] [CrossRef]
- Kadota, K.; Furukawa, R.; Shirakawa, Y.; Shimosaka, A.; Hidaka, J. Effect of surface properties of calcium carbonate on aggregation process investigated by molecular dynamics simulation. J. Mater. Sci. 2013, 49, 1724–1733. [Google Scholar] [CrossRef]
- Post, J.E.; Bish, D.L.; Heaney, P.J. Synchrotron powder X-ray diffraction study of the structure and dehydration behavior of sepiolite. Am. Miner. 2007, 92, 91–97. [Google Scholar] [CrossRef]
- Brovchenko, I.; Geiger, A.; Oleinikova, A. Water in nanopores: II. The liquid–vapour phase transition near hydrophobic surfaces. J. Phys. Condens. Matter 2004, 16, S5345–S5370. [Google Scholar] [CrossRef]
- Zhou, J.; Lu, X.; Boek, E.S. Confined water in tunnel nanopores of sepiolite: Insights from molecular simulations. Am. Miner. 2016, 101, 713–718. [Google Scholar] [CrossRef]
- Ockwig, N.W.; Greathouse, J.A.; Durkin, J.S.; Cygan, R.T.; Daemen, L.L.; Nenoff, T.M. Nanoconfined Water in Magnesium-Rich 2:1 Phyllosilicates. J. Am. Chem. Soc. 2009, 131, 8155–8162. [Google Scholar] [CrossRef] [PubMed]
- Giustetto, R.; Levy, D.; Wahyudi, O.; Ricchiardi, G.; Vitillo, J.G. Crystal structure refinement of a sepiolite/indigo Maya Blue pigment using molecular modelling and synchrotron diffraction. Eur. J. Miner. 2011, 23, 449–466. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | Alkanes | Number of Alkanes | Density of Alkanes (g/cm3) | Number of Total Atoms |
|---|---|---|---|---|
| Pal/C12 | Dodecane | 30 | 0.75 | 3336 |
| Pal/C14 | Tetradecane | 30 | 0.75 | 3516 |
| Pal/C16 | Hexadecane | 30 | 0.75 | 3696 |
| Pal/C18 | Octadecane | 30 | 0.75 | 3876 |
| Scheme | Etotal | Epal | Ealkane | △E | Ebind |
|---|---|---|---|---|---|
| Pal/C12 | −380,621.34 | −380,871.75 | 616.13 | −365.72 | 365.72 |
| Pal/C14 | −381,071.52 | −381,017.62 | 154.27 | −208.18 | 208.18 |
| Pal/C16 | −380,805.33 | −380,783.75 | 318.03 | −339.61 | 339.61 |
| Pal/C18 | −381,207.72 | −380,957.77 | 2.64 | −252.59 | 252.59 |
| Systems | Einter (kcal/mol) | Contributions to Einter (kcal/mol) | |
|---|---|---|---|
| EvdW | Eelect | ||
| Pal/C12 | −278.23 | −301.83 | 23.60 |
| Pal/C14 | −406.04 | −386.53 | −19.51 |
| Pal/C16 | −491.64 | −488.17 | −3.47 |
| Pal/C18 | −606.81 | −570.37 | −36.43 |
| Systems | Evalence (kcal/mol) | Contributions to Enon-bond (kcal/mol) | Enon-bond (kcal/mol) | Etotal (kcal/mol) | |
|---|---|---|---|---|---|
| EvdW | Eelect | ||||
| Pal/C12 | 31,880.02 | 19,245.89 | −431,743.89 | −412,498 | −380,679.12 |
| Pal/C14 | 32,259.22 | 19,357.32 | −432,677.23 | −413,319.92 | −381,124.73 |
| Pal/C16 | 31,939.41 | 19,064.11 | −431,586.91 | −412,522.79 | −380,648.69 |
| Pal/C18 | 31,985.38 | 19,171.35 | −432,020.58 | −412,849.23 | −380,932.99 |
| Systems | Y = a + b*x | D (Å2/ps) | |
|---|---|---|---|
| a | b | b/6 | |
| Pal/C12 | 14.28218 ± 0.37 | 0.20993 ± 1.44 × 10−3 | 0.03499 ± 2.41 × 10−4 |
| Pal/C14 | 11.09352 ± 0.24 | 0.07525 ± 9.39 × 10−4 | 0.01254 ± 1.57 × 10−4 |
| Pal/C16 | 7.48414 ± 0.18785 | 0.0257 ± 7.28 × 10−4 | 0.00428 ± 1.21 × 10−4 |
| Pal/C18 | 8.0188 ± 0.17 | 0.03879 ± 6.67 × 10−4 | 0.00646 ± 1.11 × 10−4 |
| Systems | Etotal | Ews | Ealkane | △E | Ebind |
| Pal/C12 | 1045.94 | 395.97 | 616.13 | 33.84 | −33.84 |
| Pal/C14 | 578.06 | 401.28 | 154.27 | 22.50 | −22.50 |
| Pal/C16 | 822.52 | 468.67 | 318.03 | 35.82 | −35.82 |
| Pal/C18 | 366.56 | 350.48 | 2.64 | 13.45 | −13.45 |
| Systems | Etotal | Ews | Ep | △E | Ebind |
|---|---|---|---|---|---|
| Pal/C12 | −380,886.28 | 395.97 | −377,686.59 | −3595.65 | 3595.65 |
| Pal/C14 | −381,017.62 | 401.28 | −377,804.38 | −3614.53 | 3614.53 |
| Pal/C16 | −380,698.07 | 468.67 | −377,259.93 | −3906.81 | 3906.81 |
| Pal/C18 | −380,898.11 | 350.48 | −377,857.79 | −3390.79 | 3390.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Yang, L.; Wang, Y.; Wu, H.; Cai, J.; Xu, S. Molecular Dynamics Simulation on the Interaction between Palygorskite Coating and Linear Chain Alkane Base Lubricant. Coatings 2021, 11, 286. https://doi.org/10.3390/coatings11030286
Zhang J, Yang L, Wang Y, Wu H, Cai J, Xu S. Molecular Dynamics Simulation on the Interaction between Palygorskite Coating and Linear Chain Alkane Base Lubricant. Coatings. 2021; 11(3):286. https://doi.org/10.3390/coatings11030286
Chicago/Turabian StyleZhang, Jin, Lv Yang, Yue Wang, Huaichao Wu, Jiabin Cai, and Shusheng Xu. 2021. "Molecular Dynamics Simulation on the Interaction between Palygorskite Coating and Linear Chain Alkane Base Lubricant" Coatings 11, no. 3: 286. https://doi.org/10.3390/coatings11030286