
Insight into the Desolvation of Organic Electrolyte Cations with Propylene Carbonate as a Solvent in Flat Pores: A First-Principles Calculation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Calculation Method
3. Results and Discussion
3.1. Reaction Principle
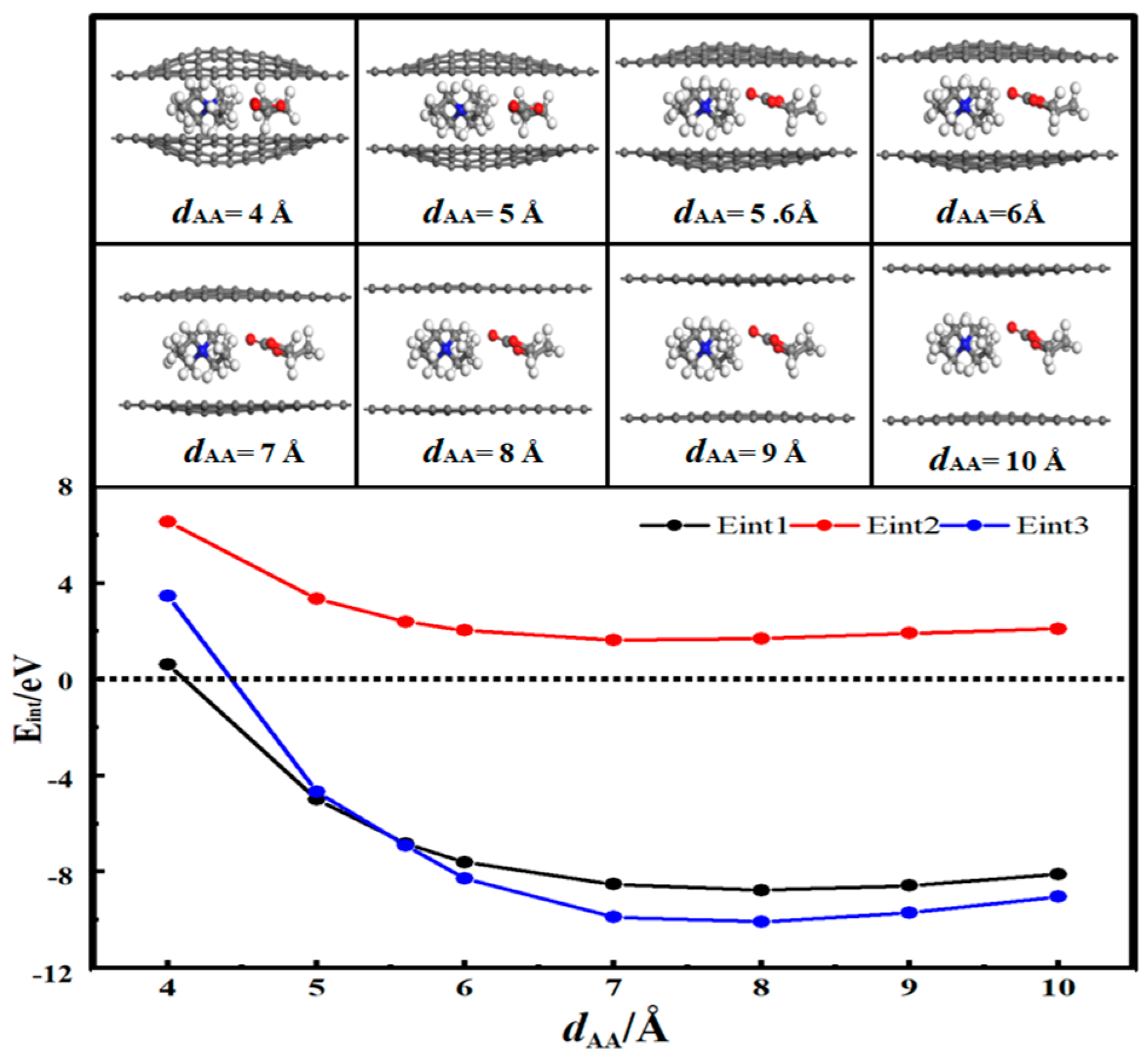
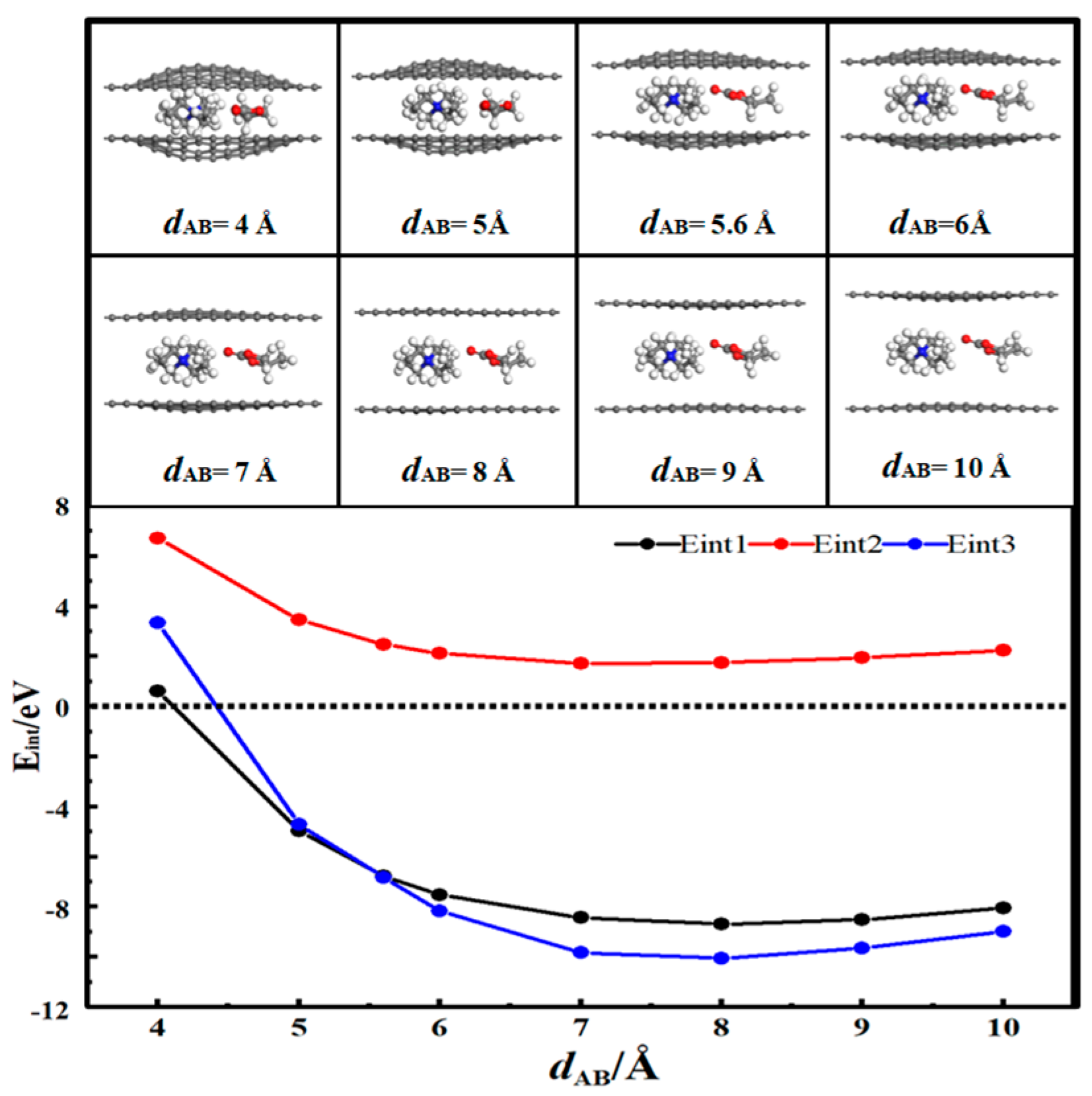
3.2. Desolvation of Li+ Complexes
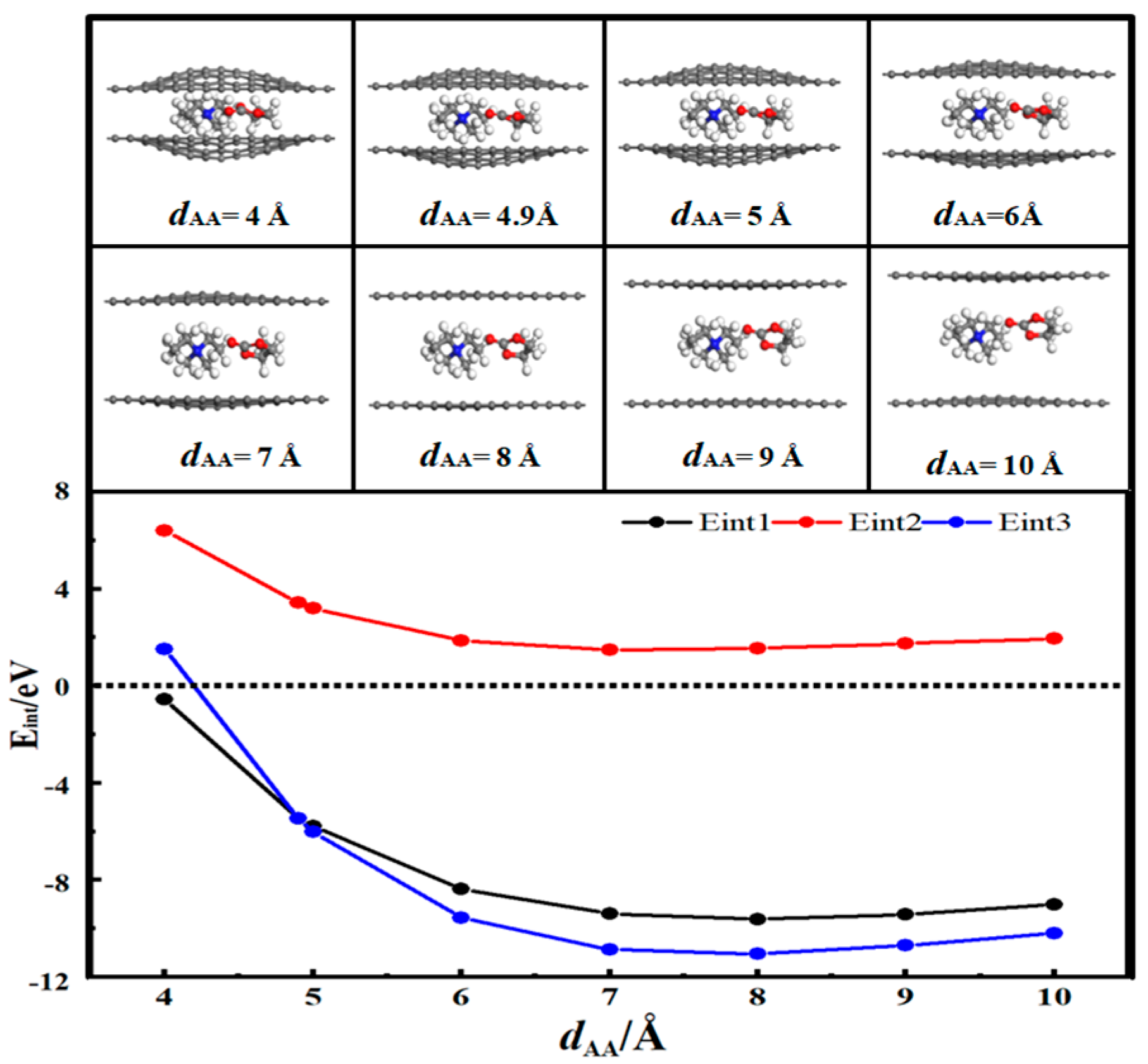
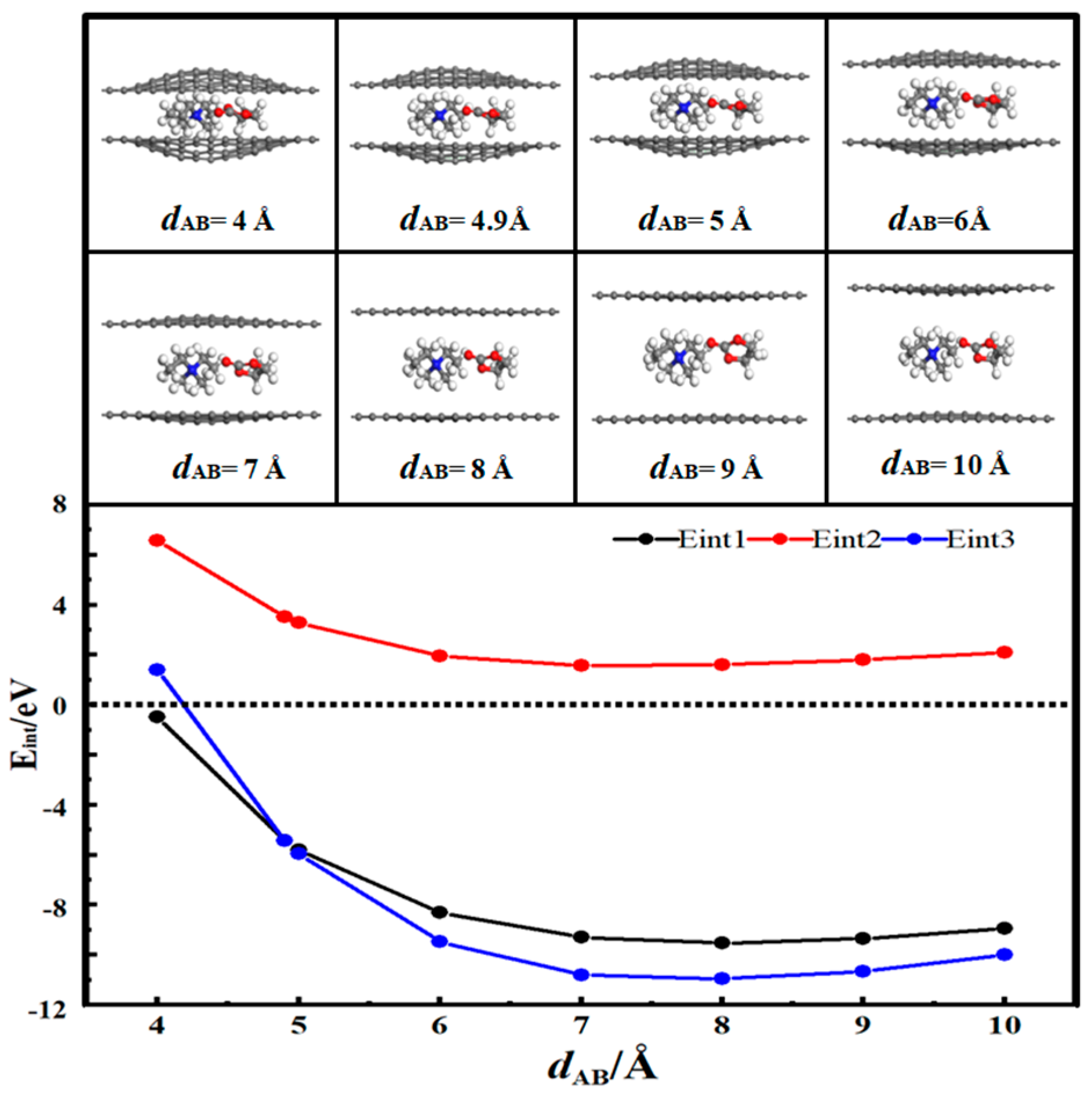
3.3. Desolvation of TEA+ Complexes
3.4. Desolvation of TEMA+ Complexes
3.5. Desolvation of SBP+ Complexes
3.6. Critical Size Analysis for Different Ionic Desolvation Processes
4. Conclusions
- In solution, the bilayer graphene system showed a fully desolvated pore size of 5.4 Å for [Li(PC)]+ and a partially desolvated pore size range from 5.4 to 5.5 Å. The fully desolvated pore size of [TEA(PC)]+ in bilayer graphene was 5.6 Å. The fully desolvated pore size of [TEMA(PC)]+ in bilayer graphene was 4.9 Å. The fully desolvated pore size of [SBP(PC)]+ in bilayer graphene was 4.8 Å.
- The relationship between the desolvation sizes of the four cations was TEA+ > Li+ > TEMA+ > SBP+.
- The three quaternary ammonium cations showed a positive trend in the complete desolvation size, with an increase in the ionic radius and an inverse trend in the size of the electric capacity, which is crucial for the development and application of supercapacitors that are used as an auxiliary power source in electric vehicles.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.; Zhang, Z.; Jiao, Y.; Yang, H.; Li, Y.; Zhang, J.; Gao, P. The grapheme/lanthanum oxide nanocomposites as electrode materials of supercapacitor. J. Power Sources 2019, 419, 99–105. [Google Scholar] [CrossRef]
- Scarpa, D.; Cirillo, C.; Ponticorvo, E.; Cirillo, C.; Attanasio, C.; Iuliano, M.; Sarno, M. Iron Selenide Particles for High-Performance Supercapacitors. Materials 2023, 16, 5309. [Google Scholar] [CrossRef]
- Liu, F.; Yang, S.; Zhang, X.; Tang, S.; Xia, Y. Insight into the Desolvation of Quaternary Ammonium Cation with Acetonitrile as a Solvent in Hydroxyl-Flat Pores: A First-Principles Calculation. Materials 2023, 16, 3858. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Sharma, K.; Arora, A.; Tripathi, S.K. Review of supercapacitors: Materials and devices. J. Energy Storage 2019, 21, 801–825. [Google Scholar]
- Zhong, C.; Deng, Y.; Hu, W. A review of electrolyte materials and compositions for electrochemical supercapacitors. Chem. Soc. Rev. 2015, 44, 7484–7539. [Google Scholar] [CrossRef]
- Beguin, F.; Presser, V.; Balduccl, A. Carbons and electrolytes for advanced supercapacitors. Adv. Mater. 2014, 26, 2219–2251. [Google Scholar] [CrossRef]
- Mayer, C.J.; Geim, K.A.; Katanelson, I.M. The structure of suspended grapheme sheets. Nat. Mater. 2007, 446, 60–63. [Google Scholar]
- Novoselov, S.K.; Geim, K.A.; Morozov, V.S. Two-dimensional gas of massless dirac fermions in grapheme. Nat. Mater. 2005, 438, 197–200. [Google Scholar]
- Fong, R.; Von, S.U.; Dahn, J.R. Studies of Lithium Intercalation into Carbons Using Nonaqueous Electrochemical Cells. J. Electro. Soc. 1990, 137, 2009. [Google Scholar] [CrossRef]
- Besenhard, J.O.; Winter, M.; Yang, J.; Biberacher, W. Filming mechanism of lithium-carbon anodes in organic and inorganic electrolytes. J. Power Sources 1995, 54, 228. [Google Scholar] [CrossRef]
- Wang, H.; Yoshio, M. Effect of cation on the performance of AC/graphite capacitor. Electro. Com. 2008, 10, 382–386. [Google Scholar] [CrossRef]
- Liang, C.; Guo, S.S.; Jie, S. Ion sieving in graphene oxide membranes via cationic control of interlayer spacing. Nature 2017, 550, 380–383. [Google Scholar]
- Zhao, P.; Su, Y.; Zhang, Y.; Li, S.J.; Chen, G. CO catalytic oxidation on iron-embedded hexagonal boron nitride sheet. Chem. Phys. Lett. 2011, 515, 159–162. [Google Scholar] [CrossRef]
- Mattsson, A.E.; Schultz, P.A.; Desjarlais, M.P.; Mattsson, T.R.; Leung, K. Designing meaningful density functional theory calculations in materials science-a primer. Modell. Simul. Mater. Sci. Eng. 2004, 13, 1–31. [Google Scholar] [CrossRef]
- Rajagopal, A.K.; Callaway, J. Inhomogeneous Electron Gas. Phys. Rev. B 1973, 7, 1912–1919. [Google Scholar] [CrossRef]
- Seifert, G.; Porezag, D.; Frauenheim, T. Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA Scheme. Int. J. Quantum. Chem. 1996, 58, 185–192. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.T.; Frauenheim, S.; Su, H.; Seifert, G. Self-consistent charge density functional tight-binding method for simulation of complex material properties. Phys. Rev. B 1998, 58, 7260. [Google Scholar] [CrossRef]
- Seifert, G. Tight-binding density functional theory: An approximate Kohn-Sham DFT scheme. J. Phys. Chem. A 2007, 111, 5609. [Google Scholar] [CrossRef]
- Segall, D.M.; Lindan, D.J.P.; Probert, J.M.; Pickard, J.C.; Hasnip, J.P.; Clark, J.S.; Payne, C.M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter. 2002, 14, 2717. [Google Scholar] [CrossRef]
- Yang, S.B.; Liu, X.L.; Zhang, X.; Tang, S.W. Insights into the effect of hydroxyl-, epoxy-, and carboxyl-pores on the desolvation of K+ with water as a solvent: A first-principles study. J. Phys. Condens. Matter. 2021, 33, 445201. [Google Scholar] [CrossRef]
- Wu, S.; Yan, G.; Cheng, X. First-principles investigation of the superconducting properties of thallium sulfide. Phys. C 2019, 562, 1–6. [Google Scholar] [CrossRef]
- Schira, R.; Latouche, C. DFT and hybrid-DFT calculations on the electronic Properties of vanadate materials: Theory meets experiments. New J. Chem. 2020, 44, 11602. [Google Scholar] [CrossRef]
- Monkhorst, J.H.; Pack, D.J. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Tonel, M.Z.; Zanella, I.; Fagan, S.B. Theoretical study of small aromatic molecules adsorbed in pristineand functionalized graphene. J. Mol. Model. 2021, 27, 193. [Google Scholar] [CrossRef] [PubMed]
- Calborean, A.; Buimaga⁃Iarinca, L.; Graur, F. DFT charge transfer of hybrid molecular ferrocene/Si structures. Phys. Scr. 2015, 90, 055803. [Google Scholar] [CrossRef]
- Rabone, J.; Uffelen, P.V. DFT-based Metadynamics simulation of proton diffusion in tetragonal zirconia at 1500 K. J. Nucl. Mater. 2015, 459, 30–36. [Google Scholar] [CrossRef]
- Dolgonos, G.; Aradi, B.; Moreira, N.H.; Frauenheim, T. An Improved Self-Consistent-Charge Density-Functional Tight-Binding (SCC-DFTB) Set of Parameters for Simulation of Bulk and Molecular Systems Involving Titanium. J. Chem. Theory Comput. 2010, 6, 266. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, S.B.; Shan, X.Y. Insights into the effect of the interlayer spacings of bilayer graphene on the desolvation of H+, Li+, Na+, and K+ ions with water as a solvent: A first-principles study. Phy. Chem. 2019, 21, 23697. [Google Scholar] [CrossRef]
- Ivanova, S.; Laguna, H.O.; Centeno, A.M. Microprocess technology for hydrogen purification. Renew. Hydrogen. Technol. 2013, 10, 225–243. [Google Scholar]
- Rufford, E.T.; Hulicova-Jurcakova, D.; Zhu, H.Z.; Lus, G.Q. Empirical Analysis of the Contributions of Mesopores and Micropores to the Double-Layer Capacitance of Carbons. J. Phys. Chem. C 2009, 113, 19335. [Google Scholar] [CrossRef]
- Chmiola, J.; Largeot, C.; Taberna, L.P.; Simon, P.; Gogotsi, Y. Desolvation of Ions in Subnanometer Pores and Its Effect on Capacitance and Double-Layer Theory. Angew. Chem. Int. Ed. 2008, 47, 3392. [Google Scholar] [CrossRef]
- Nono, Y.; Kouzu, M.; Takei, K.; Chiba, K.; Sato, Y. EDLC Performance of Various Activated Carbons in SpiroType Quaternary Ammonium Salt Electrolyte Solutions. Electrochemistry 2010, 78, 336–338. [Google Scholar] [CrossRef] [Green Version]
- Chiba, K.; Ueda, T.; Yamamoto, H. Performance of electrolyte composed of spiro-type quaternary ammonium salt and electric double-layer capacitor using it. Electrochemistry 2007, 75, 664–667. [Google Scholar] [CrossRef] [Green Version]
- Janes, A.; Lust, E. Use of organic esters as co-solvents for electrical double layer capacitors with low temperature performance. J. Electro. Chem. 2006, 588, 285–295. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Yang, S.; Zhang, X.; Tang, S.; Wei, S. Insight into the Desolvation of Organic Electrolyte Cations with Propylene Carbonate as a Solvent in Flat Pores: A First-Principles Calculation. Coatings 2023, 13, 1384. https://doi.org/10.3390/coatings13081384
Liu F, Yang S, Zhang X, Tang S, Wei S. Insight into the Desolvation of Organic Electrolyte Cations with Propylene Carbonate as a Solvent in Flat Pores: A First-Principles Calculation. Coatings. 2023; 13(8):1384. https://doi.org/10.3390/coatings13081384
Chicago/Turabian StyleLiu, Fudong, Shaobin Yang, Xu Zhang, Shuwei Tang, and Shuang Wei. 2023. "Insight into the Desolvation of Organic Electrolyte Cations with Propylene Carbonate as a Solvent in Flat Pores: A First-Principles Calculation" Coatings 13, no. 8: 1384. https://doi.org/10.3390/coatings13081384