1. Introduction
In recent years, progress made in the area of materials science has opened great opportunities for the use of nanoparticles in biotechnological applications. Inorganic materials are one of the most investigated types of materials for their properties and offer tremendous opportunities in chemistry, physics and biology. These materials are considered promising candidates in the development of newly nanostructured materials and devices with controllable physicochemical and biological properties. Moreover, in recent decades, important advances have been made in the fabrication, characterization, and tailoring of the properties of nanoparticles, generating valuable solutions for numerous biomedical applications [
1,
2,
3]. Among inorganic materials, magnetic particles have been intensively studied since their introduction in the 1970s for their exquisite properties for applications in medicine and biology for magnetic targeting (targeted drug delivery, targeted gene therapy, targeted radionuclides), magnetic resonance imaging (MRI), and immunoassay applications (gene therapy, cell labeling, ribonucleic acid (RNA) and deoxyribonucleic acid (DNA) purification, cell separation, cell purification), as well as hyperthermia generation [
4,
5,
6,
7,
8,
9,
10]. Therefore, these materials cover the entire processes involved in current medical practices (i.e., diagnostics, therapy, and treatment). The use of small-scale iron oxide has been common practice approximately 40 years, but in recent decades a considerable deal of attention has been focused towards the study of several types of iron oxide nanoparticles, magnetite (Fe
3O
4), and maghemite (γ-Fe
2O
3), due to the special properties that they exhibit at the nanometric scale [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10]. Magnetite is an iron oxide with a cubic inverse spinel in which oxygen forms a closed face-centered cubic (fcc) structure and Fe cations occupy the interstitial (tetrahedral and octahedral) sites. Due to this structure, the electrons can hop between the Fe
2+ and Fe
3+ ions in the octahedral sites, even at room temperature, making magnetite an important class of half-metallic materials [
11]. Furthermore, using a convenient surface coating, magnetic nanoparticles can be dispersed using certain solvents and can be used to obtain homogenous suspensions, designated as ferrofluids [
12,
13,
14,
15]. In recent decades, biocompatible aqueous-based magnetic fluids have attracted a great deal of attention in the development of novel biomedical applications such as magnetic resonance imaging, magnetic targeted drug delivery, magnetic intracellular hyperthermia, magnetic cell separation, and alternating current (AC) magnetic field-assisted cancer therapy, etc. [
2,
12,
13,
14,
15,
16,
17]. In order to obtain stable magnetite magnetic fluids, usually, a biocompatible surface coating, commonly a natural polymer, is used. Polysaccharides are common natural polymers, formed of repeated monosaccharides that have numerous reactive groups, such as hydroxyl (–OH) amino (–NH
2) groups. These materials are often used as coatings for magnetic nanoparticles due to their antifouling and biocompatible properties. Moreover, they are also generally appreciated due to their versatility for further modifications. Recent studies have highlighted that polysaccharides could be successfully used as anti-fouling coatings for magnetic resonance imaging agents in the reduction of protein adsorption and for improving the biocompatibility of magnetic nanoparticles [
2,
7,
18,
19,
20,
21,
22]. Dextran is a natural glucose-based polysaccharide and is one of the most used polymers used in the coating of magnetic nanoparticles [
18,
19,
20]. This natural polymer is preferred due to its exquisite properties of solubility, low toxicity, biocompatibility, and its high affinity to iron oxide nanoparticles [
6,
22]. The prevention of non-specific protein adsorption is of utmost importance in the development of biocompatible material agents for medical implants, diagnostics, and therapeutics [
2,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22]. In this context, preliminary studies regarding the development of magnetite magnetic fluids stabilized using dextran have been reported since 1978 by Syusaburo and Masalatsu [
18], and later studied by different research teams (S19 and S20). Moreover, several nanoparticles of this class are now approved by the United States Food and Drug Administration (FDA) and are used successfully in the medical field. Feridex I.V. (ferumoxides) is the first nanoparticle-based iron oxide imaging agent that has been approved by FDA (it was approved in 1996) and it is used in the detection of liver lesions [
7]. Since then, other versions, like Combidex (ferumoxtran-10) and Feraheme (ferumoxytol) have been approved to be used in human diagnostics and treatment. Due to their proven biocompatibility, dextran-coated iron oxide nanoparticles are already recognized for their use in the development of multifunctional imaging agents [
19,
20,
21]. Recently, in order to improve their efficiency, researchers have focused on the development of thin layers of magnetic nanoparticles. Processing the material into a thin layer improves the treatment efficiency and allows the obtainment of a homogeneous coating with a controlled release of an active component, having the same resemblance as the starting material. In this context, the aim of this study was the obtainment of stable dextran-coated iron oxide nanoparticles solutions (D-MNPs) and using them in the development of uniform and homogenous thin layers for biomedical applications. Complementary studies such as dynamic light scattering (DLS), zeta potential (ZP), TEM, XRD and ultrasound measurements were performed on the D-MNP suspensions that were used to make the coatings. The morphology, uniformity, and chemical compositions of the obtained layers were also studied by scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS). Furthermore, cytotoxicity and cell viability studies were performed on both the D-MNP suspensions and the obtained layer.
2. Materials and Methods
2.1. Materials Reagents
Ferric chloride hexahidrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), natrium hydroxide (NaOH), dextran (H(C6H10O5)xOH) with a molecular weight of ~40,000, perchloric acid (HClO4), and hydrochloric acid (HCl) were acquired from Merck (Bucharest, Romania). Deionized water was also used. A silicon substrate, using the spin-coating procedure, was purchased from Sil’tronix Silicon Technologies (Archamps, France).
2.2. Synthesis of D-MNPs
Chloride hexahydrate (FeCl3·6H2O) dissolved in water and ferrous chloride tetrahydrate (FeCl2·4H2O) in 2 M HCl were mixed at room temperature. The molar ratio of [Fe3+]/[Fe2+] was 2. The mixture was added drop by drop into the dextran solution (30% w/v) and 400 mL NaOH (2 M) solution at 90 °C under continuous stirring for about 45 min. The resulting solution was continuously stirred (200 rpm) for 1 h at 90 °C. A 5 M NaOH solution was added dropwise to obtain a pH equal to 12. The black precipitate was treated repeatedly with a 3 M perchloric acid solution (400 mL). The [Fe2+]/[Fe3+] ratio in final suspension was about 0.05. The last separation was performed by centrifugation at 16,000 rpm for 1 h. Finally, the particles were re-dispersed into a dextran solution. Finally, the dextran-coated iron oxide nanoparticle (D-MNP) solutions were obtained.
2.3. Thin Layer of D-MNPs
To achieve the thin layer, 0.3 mL of the D-MNP resulting solution was used. Here, 0.3 mL was pipetted using a syringe on the top of the silicon substrate. During the deposition, the silicon substrate was rotated at 1000 rpm for 40 s. The process was repeated 20 times. After each coating, the layer was dried in a nitrogen atmosphere for 1 h at 70 °C immediately after coating. After the last coating, the resulting layer was dried in a nitrogen atmosphere for 1 h at 70 °C and then treated at 100 °C under vacuum for 1 h to further densify the film.
2.4. Characterization Methods
Ultrasonic measurements were performed in a 100 mL suspension of D-MNPs, which were previously stirred for 30 min at room temperature using a magnetic stirrer (Velp, Usmate, Italy). After stopping the stirring, ultrasound pulses were sent through D-MNP suspension [
23]. The recording of digital signals from the oscilloscope at a very precise interval of 5.00 s was done using an electronic device. The evolution of the signals in time offers information both on the stability of the suspension and the attenuation of the signals in time. For signal processing, under the same experimental conditions, double distilled water was considered as the reference fluid.
Dynamic light scattering measurements (DLS) and ζ-potential evaluations were performed on a SZ-100 Nanoparticle Analyzer (25 ± 1 °C; laser wavelength 532 nm) from Horiba-Jobin Yvon (Horiba Ltd., Kyoto, Japan). The scattering angle was 173° and the primary data were obtained from the correlation function of the scattered intensity. The dispersion medium viscosity was 0.895 mPa·s. The obtained samples were diluted 10 times in ethanol before the investigations of the DLS and ζ-potential. To determine the hydrodynamic diameter (DH), the sample was illuminated with a laser source that allowed the estimation of the particle diffusion velocity. For each analyzed sample, there were three recorded determinations. The final value was determined by averaging the three measurements.
In order to evaluate the morphology of the D-MNP sample, a scanning electron microscope (SEM) with a HITACHI S4500 (Hitachi Ltd., Chiyoda, Japan) was used. A dedicated attachment to the SEM apparatus allowed us to assess the chemical composition of the D-MNP sample by energy dispersive X-ray spectroscopy (EDS) at 20 kV. The morphology of the samples was investigated by transmission electron microscopy using a CM 20 (Philips-FEI, Hillsboro, OR, USA) transmission electron microscope equipped with a Lab6 filament (Agar Scientific Ltd., Stansted, UK) operating at 200 kV. The XRD patterns of the synthesized D-MNP samples were recorded with a Bruker D8 Advance diffractometer (Bruker, Karlsruhe, Germany), using Cu Kα (λ = 1.5418 Å) radiation in the 2θ ranging from 20° to 70° and a high efficiency LynxEye™ linear detector (Bruker, Karlsruhe, Germany). The crystallite sizes of the D-MNP samples were appraised from the X-ray line broadening using Scherrer formula:
where
d is the mean size of the ordered (crystalline) domains, which may be smaller or equal to the grain size,
K is a dimensionless shape factor, with a value close to unity. The shape factor has a typical value of about 0.9, but varies with the actual shape of the crystallite, λ is the X-ray wavelength, β is the line broadening at half the maximum intensity (FWHM) after subtracting the instrumental line broadening, in radians, and θ is the Bragg angle.
2.5. Hela Cell Viability Assays
The cytotoxicity assays of the samples were performed using the HeLa cell line Sigma-Aldrich Corp., St. Louis, MO, USA). In order to study the in vitro interaction of the HeLa cells with the samples, two-time intervals were chosen (24 and 48 h). The HeLa cells were trypsinized, counted, and seeded into 24-well plates at a density of 5 × 104 cells per well. The plates were incubated in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich Corp., St. Louis, MO, USA), supplemented with 10% fetal bovine serum (Sigma-Aldrich Corp., St. Louis, MO, USA), at 37 °C, in a humid atmosphere of 5% CO2 for 24 h. To assess the cytotoxicity of the D-MNP solutions, the cells were treated with 100 µg/mL of solution. The treated cells were then kept for 24 and 48 h at 37 °C, in a 5% CO2 atmosphere, and a quantitative assay of the HeLa cell’s viability was conducted using the conventional 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. The results were interpreted as percentages of the viable cells treated with D-MNP solution relative to the viability of untreated cells (control, 100%). In the case of the D-MNP layers, the HeLa cells previously cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich Corp., St. Louis, MO, USA), supplemented with 10% fetal bovine serum (Sigma-Aldrich Corp., St. Louis, MO, USA), at 37 °C, in a humid atmosphere of 5% CO2 for 24 h, were incubated with the D-MNP layers for 24 and 48 h. After 24 and 48 h of incubation, the cell viability was evaluated using the MTT reduction assay. Furthermore, the morphology and the cell cycle features of the HeLa cells incubated with both D-MNP suspensions and D-MNP layers were also analyzed. For that purpose, after being incubated for 24, respective to 48 h with the HeLa cell cultures, the samples were fixed using cold ethanol, stained with propidium iodide (PI), and visualized with the aid of a Leica DFC 450C fluorescence microscope (Leica Camera, Wetzlar, Germany). After growing HeLa cells in the presence of the samples for 24 and 48 h, the cell suspension was fixed in 70% ethanol for at least 30 min. After centrifugation, the cell pellet was resuspended in a 1 mL phosphate buffer containing 50 g/mL RNA-z and PI. The stained cells were analyzed at the flow cytometer within 2 h after fixation. DNA histogram deconvolution analysis was performed using FlowJo software (FlowJo v9) and the Watson model, measuring the percentage of cells in the G0/G, S, G2/M phases. The cytotoxic assays were conducted in triplicate and the results were presented as mean ± SD.
2.6. Statistical Analysis
The biological studies were carried out in triplicate. To perform the statistical analysis, a t-test and analysis of variance (ANOVA) were used. Differences between samples were considered to be significant at p < 0.05.
3. Results and Discussions
To estimate the stability of the D-MNP suspension used to obtain the thin layers, a non-destructive ultrasound-based technique was used.
The ultrasound studies were performed on the D-MNP suspension obtained by the coprecipitation method. The stability of the D-MNP suspension was studied both according to the evolution in time of the signals obtained from sending ultrasonic pulses through the studied suspension, depending on the evolution in time of the attenuation. The processing of the obtained results was performed according to the results obtained for double distilled water under the same experimental conditions. The exact determination of the speed of ultrasound in the D-MNP suspension for each signal was established according to the time delays between the first three recorded echoes and those of the reference suspension (bi-distilled water in our case). For the D-MNP suspension, the calculated velocity at 24 °C was 1508.2 m/s, while for the reference fluid it was 1494.84 m/s. This speed cannot be a reliable parameter in the characterization of the evolution of the sample as it has very little variation during the sedimentation of the particles. From the maximum amplitude of the transmitted signals compared to the recording moments, a more significant variation was determined. As can be seen in
Figure 1, there was a slow and continuous variation of amplitudes, which will be used for further processing. Moreover, during the recording (3200 s), the suspension did not precipitate. On the other hand, the commonly encountered passage of the separation surface in front of the transducers does not appear in this case. As can be seen in the figure, there is a very brief initial interval of less than 60 s, during which the amplitude increases faster, followed by more than 3000 s of slow evolution. The first transmitted echo, which is best recorded, tends slowly towards a relative amplitude which is below 1, the value which would correspond to pure water. This remark is also an indicator of the high stability of the suspension, which will be quantitatively determined in the following section.
The entire measurement period is relevant for the stability of this particular suspension. It can be observed that during this period of time, the amplitude of the first echo, which was determined with the highest precision, showed a slowly increasing value. The slope of this amplitude vs. time is related to the stability parameter, which in this case is
= 0.0000444 (1/s), with
Am being the averaged amplitude of the signals. This small value indicates an excellent stability. It is reminded here that for pure water
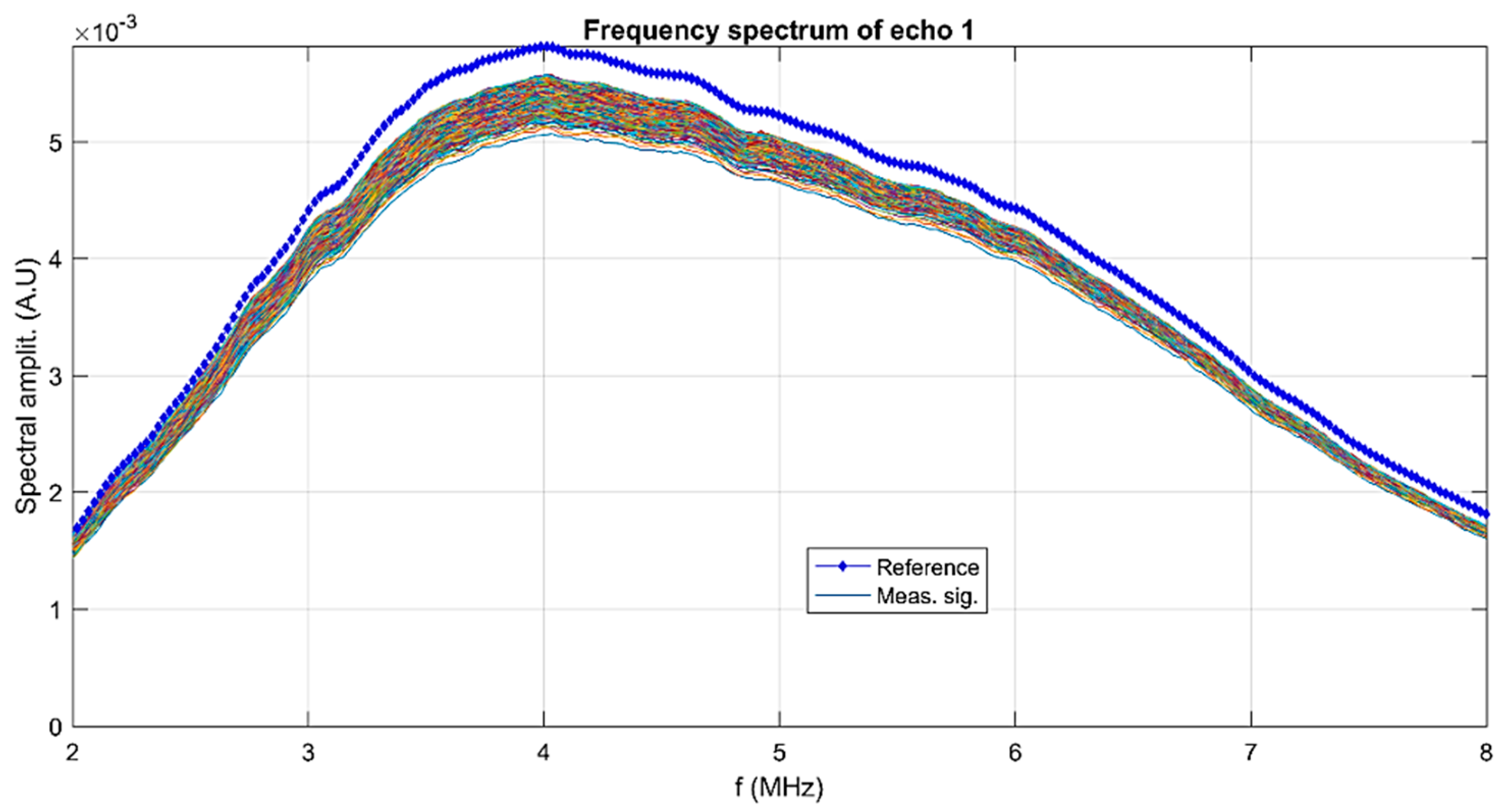
s = 0. The suspension has a typical signature in the frequency spectrum of the first transmitted echo (
Figure 2).
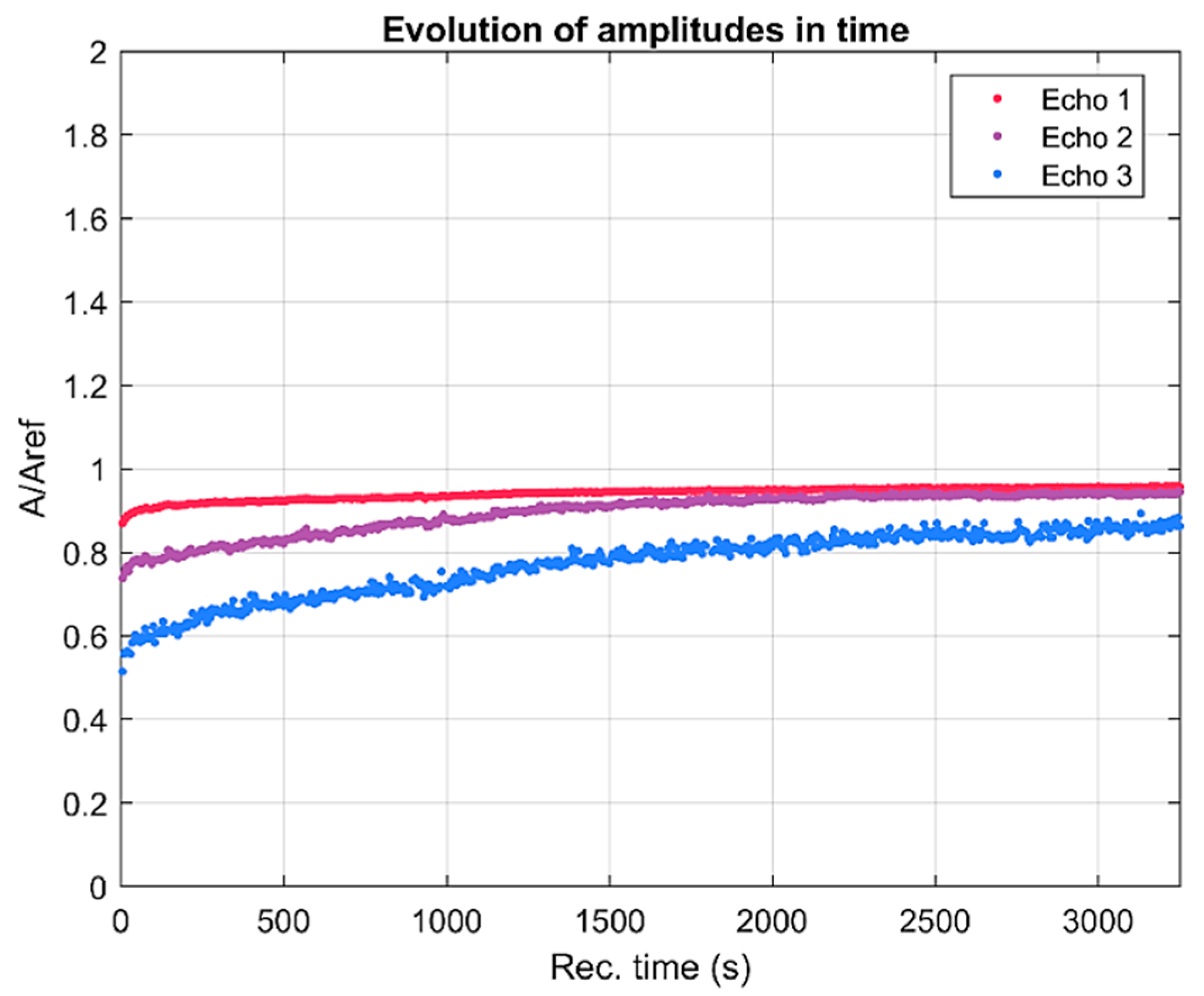
The lowest curves correspond to the first period of sedimentation and the upper ones correspond to the asymptotic sedimentation of the suspension fluid. The peak is at 4 MHz, which is a characteristic of the transducers. The spectrum for the reference fluid is indicated by blue markers. Another piece of information concerning the suspension is the spectral amplitude variation during the experiment. The recorded first echo was split into harmonic components of frequencies between 2 and 8 MHz (
Figure 3). It is interesting to point out that the component at 2 MHz tends, on average, to a relative amplitude of 1, meaning that the suspension becomes transparent to signals at this frequency. All higher frequencies exhibit a continuous increasing path for their relative amplitudes, from a minimum of 0.86 towards 0.97 during the long recording interval. The evolution for each frequency is difficult to extract from
Figure 3.
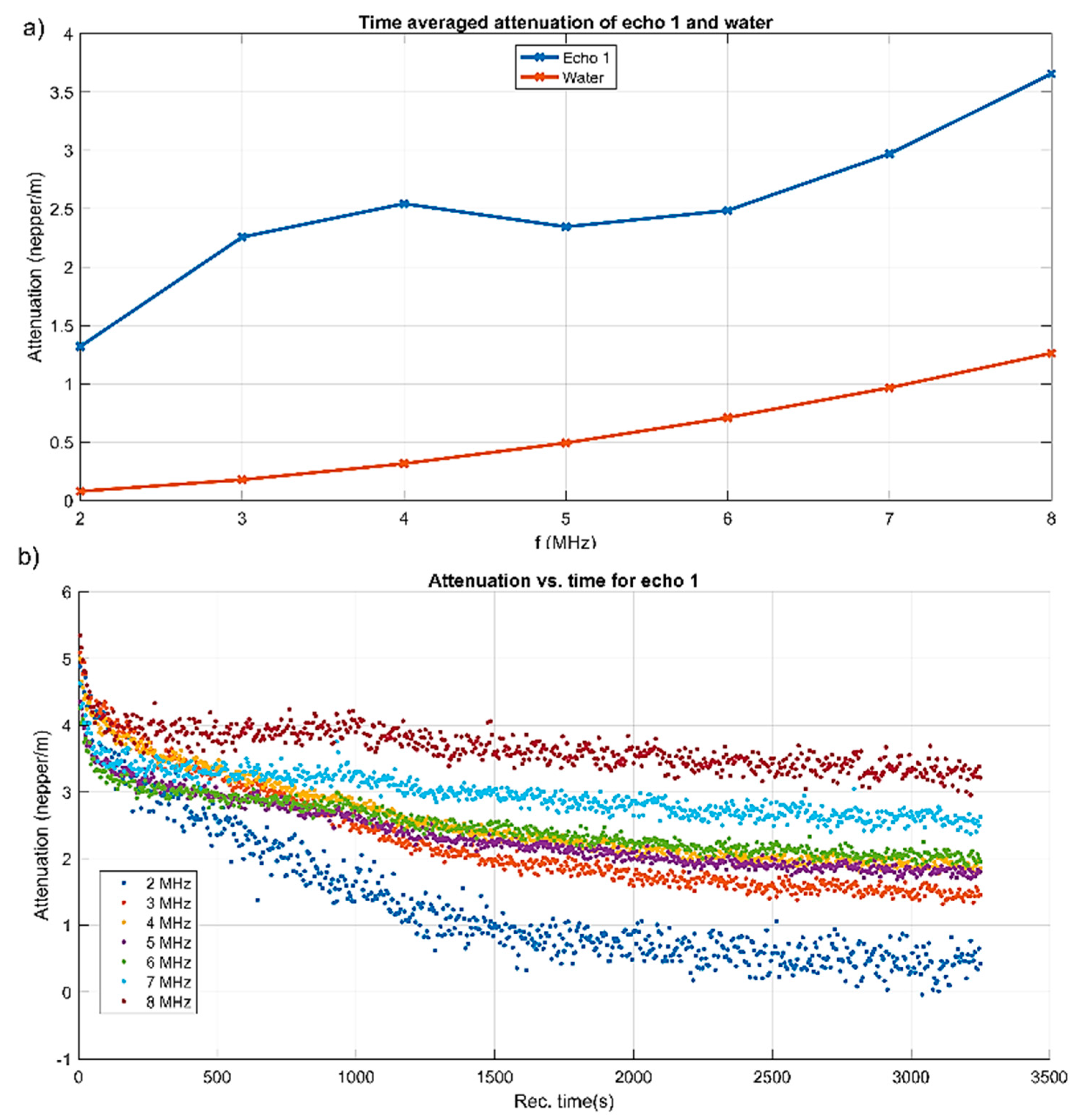
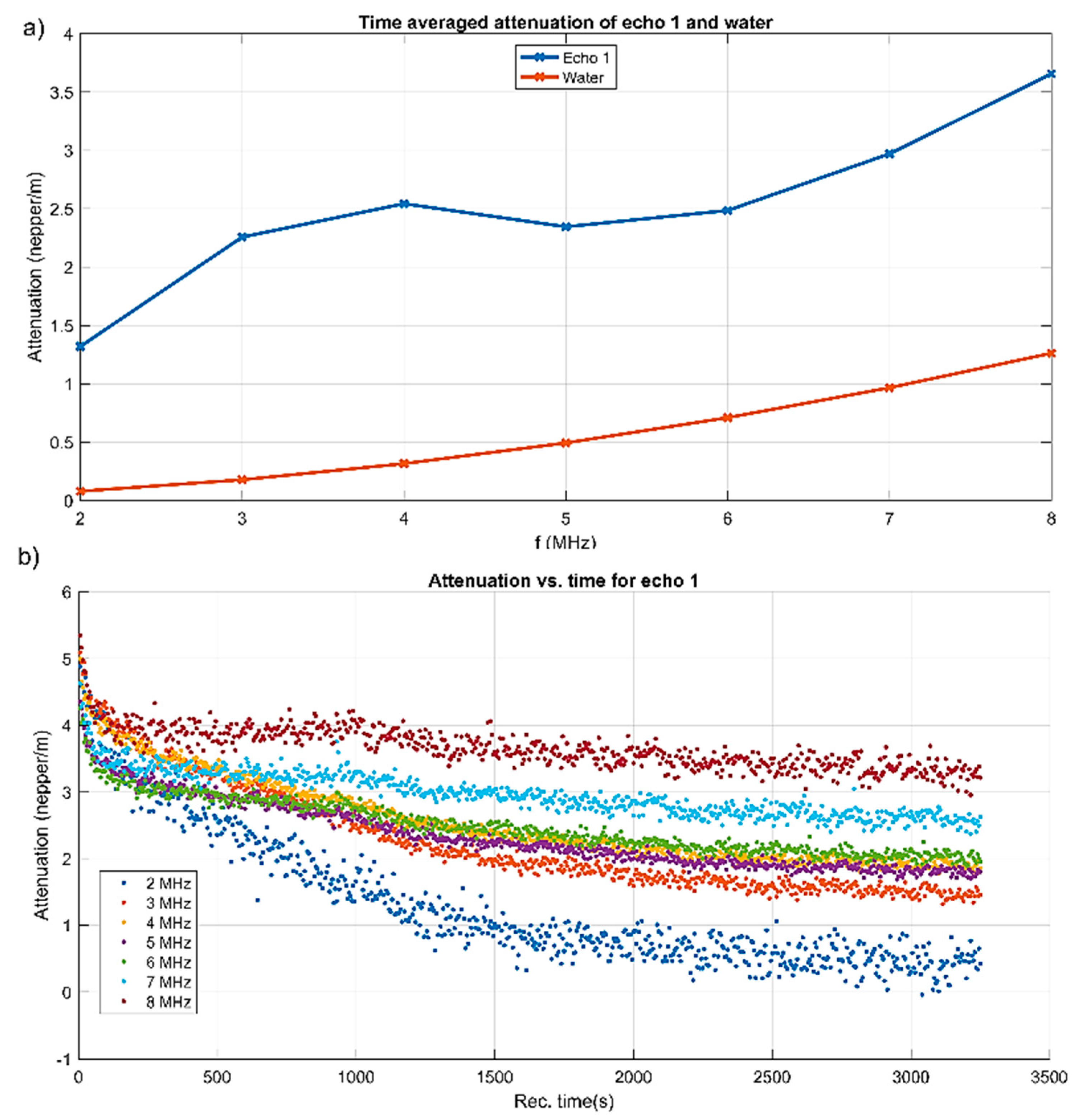
More explicit information on the attenuation dependency of frequency is given in
Figure 4a, compared to the same dependency on frequency for the reference fluid. The attenuation is considerably higher (1.3–3.6 nepper/m) for the sample compared to 0.3–1.3 nepper/m for the reference fluid. A characteristic of this suspension is the local maxima of attenuation between 3 and 4 MHz, at which acoustic energy is absorbed and scattered more than in an equivalent homogeneous fluid.
Certainly, the attenuation for each spectral component depends on the moment during the experiment, as shown in
Figure 4b. At all selected frequency components, the initial attenuation was in a narrow interval of 4–5.5 nepper/m. The components at 2 and 3 MHz kept a higher attenuation in time, which descended towards 2.6 and respectively 3.3 dB/m after 3250 s of monitoring time. The other frequencies diminished their attenuation to lower than 2 nepper/m. Only the 2 MHz component tended to a very low attenuation. As a partial conclusion, a transducer with a central frequency as low as 2 MHz would not be capable to extract the distinct features of this sample.
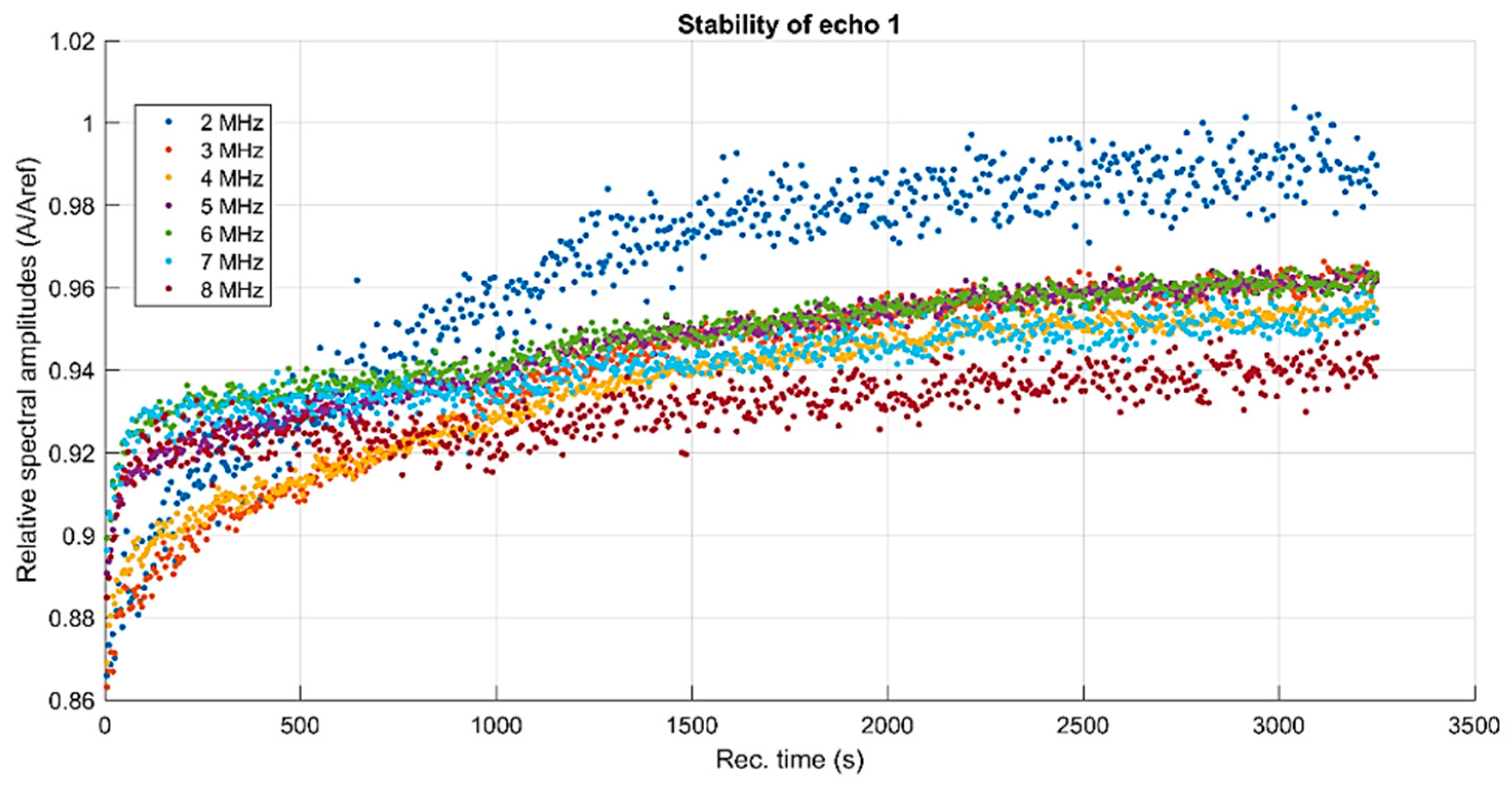
The D-MNP suspension reaches a very stable state after about 1 min and remains stable, with a maximum amplitude variation of 4%, for a very long time.
Complementary studies to investigate the stability of the suspension to be used in the production of thin layers were performed by DLS. DLS and ζ-potential studies were performed on diluted solutions while ultrasound measurements were performed on the solution used to make the thin layers.
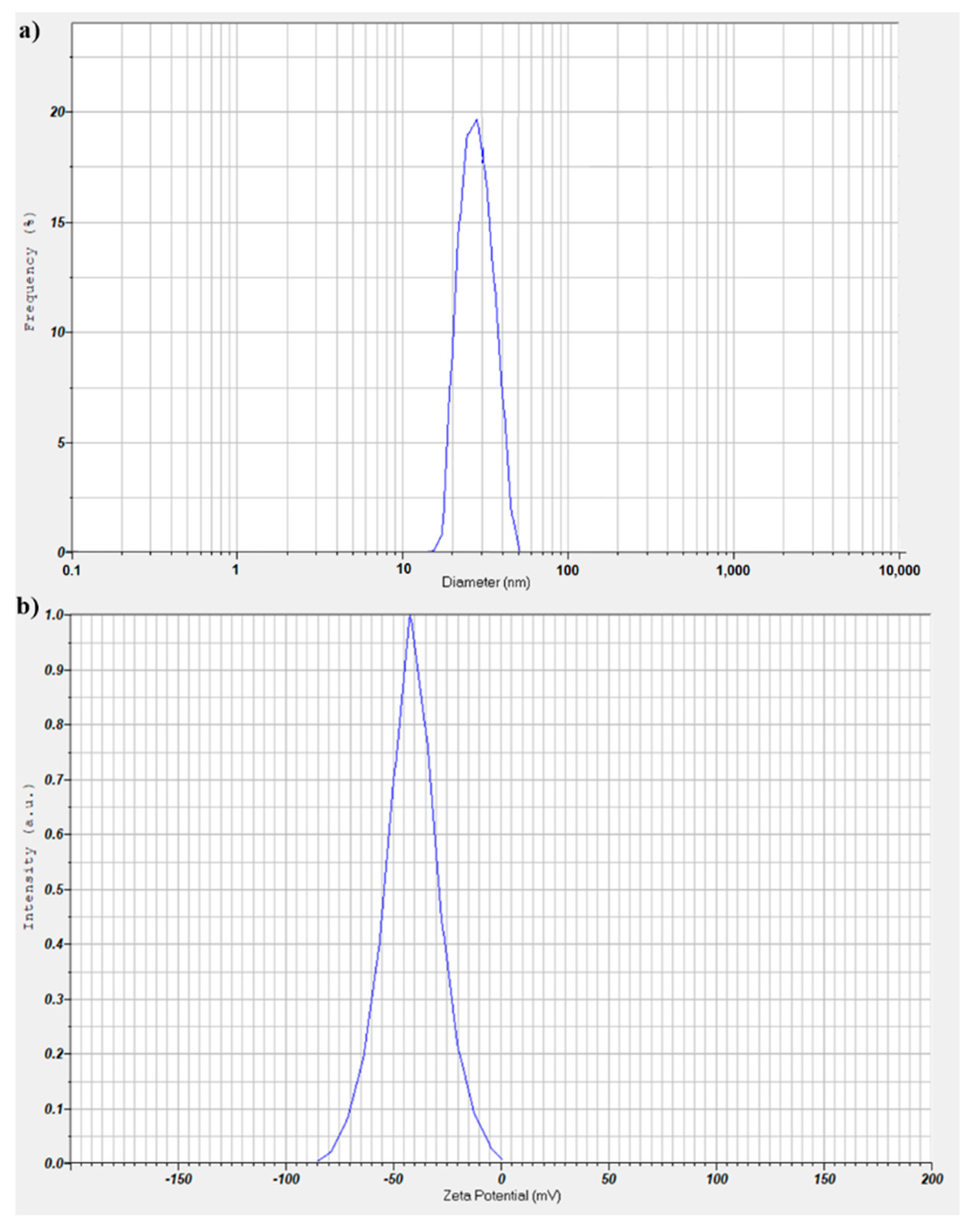
The DLS and ζ-potential investigations were performed on dilutions of the stable suspension of D-MNPs maintained under ambient conditions. The results obtained from the DLS investigations and ζ-potential are presented in
Figure 5. For the DLS and ζ-potential measurements, the suspensions of D-MNP were diluted 5 times. The average hydrodynamic size (
DH) obtained by DLS was 27.8 ± 5 nm (
Figure 5a). On the other hand, it was observed that the particles were monodispersed.
The stability of the D-NMPs suspension depends on the electrostatic repulsion. As a result, measuring the ζ-potential for D-MNP suspensions is very important. Here, the ζ-potential value for D-NMs suspensions was −44.1 mV (
Figure 5b). This value of the ζ-potential ensures an electrostatic repulsion force between particles that is large enough to prevent the attraction and collision caused by Brownian motion. The value of the ζ-potential showed that the stability of the D-MNP suspension was good.
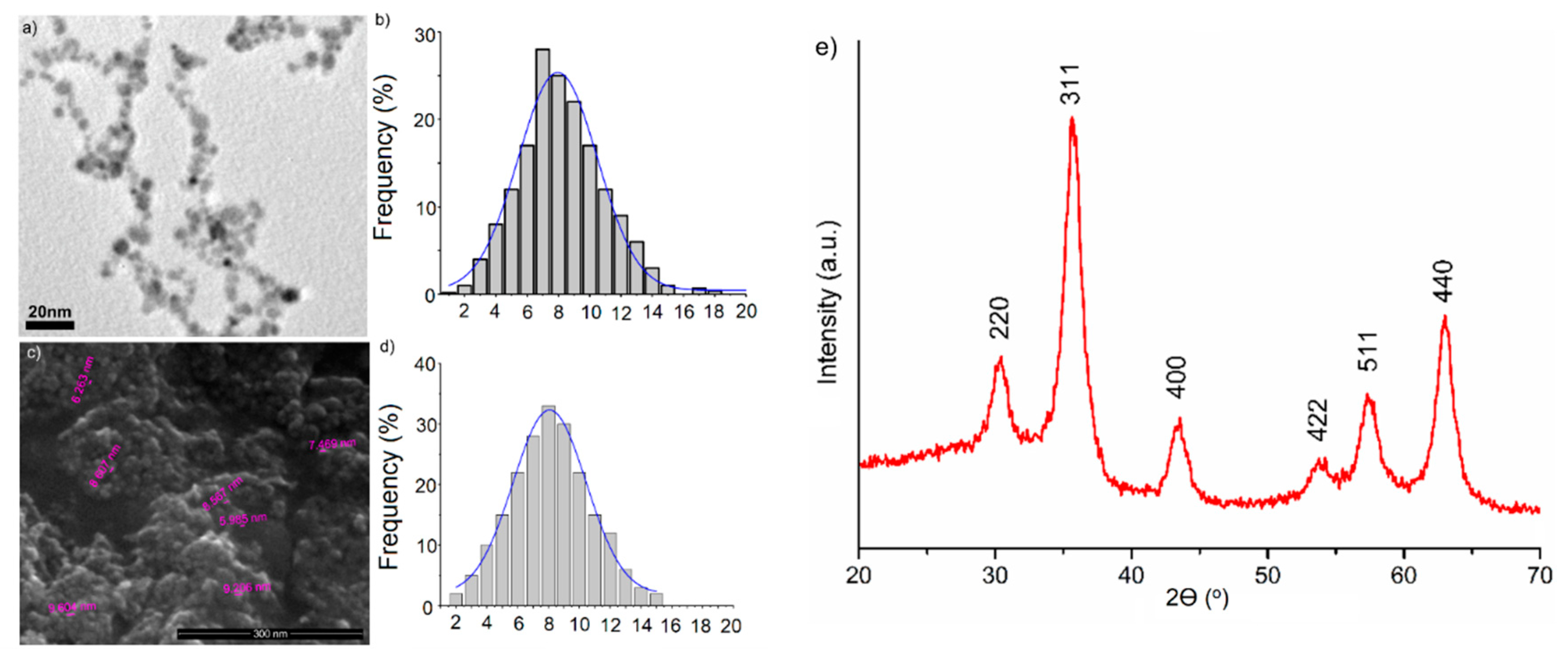
The morphology and size of D-MNPs was evaluated by TEM and SEM analysis (
Figure 6). The TEM results showed that dextran-coated iron oxide particles are spherical in shape and are dispersed (
Figure 6a). The average diameter calculated from the size distribution was 7.3 ± 0.6 nm (
Figure 6b). The size distribution was obtained after measuring about 1000 particles. A SEM micrograph of the D-MNP particles is presented in
Figure 6c. It can see that the morphology of the D-MNP particles was uniform. The particles are spherical, with nanoscale dimensions. The average diameter calculated from SEM image was 8.5 ± 0.4 nm (
Figure 6d).
In order to investigate the crystalline structure of D-MNPs, the D-MNPs suspensions were centrifuged at 12,000 rot/min and the resulting powder was dried in an oven at 80 °C. The X-ray diffractogram of the D-MNP powder after drying at 80 °C is presented in
Figure 6e. All diffraction characteristic peaks corresponding to (220), (311), (400), (511) and (440) planes were identified. The identified diffraction peaks were indexed to a face-centered cubic spinel structure (Fd3m3 space group) with a lattice parameter of
a = 8.334 Å (JCPDS card No. 39-1346). The lattice parameter of the synthesized D-MNP (
a = 8.334 Å) was in agreement with the bulk lattice parameter of maghemite (
a = 8.3474 Å). These results were in agreement with those previously reported in the literature [
8,
24,
25,
26]. Scherrer’s equation [
27] was used to calculate the crystallite size of D-MNPs from the XRD line broadening. The calculated crystallite size (
DXRD) was 7.1 ± 0.1 nm. The very good crystallinity of the D-MNPs was confirmed by the XRD patterns.
The
DXRD was in good agreement with the size of D-MNPs obtained from TEM micrographs (
DTEM). On the other hand, the
DH obtained by DLS was substantially higher than the
DTEM obtained from the TEM micrographs. This difference may be due to the fact that the TEM analysis was done on powder while the DLS studies were performed on suspensions. The difference between the TEM and DLS results may be due to the fact that TEM analysis cannot measure any particle coating agent, while DLS provides information on both the particle diameter and the molecules or ions that are attached to its surface [
28]. On the other hand, the DLS technique also measured possible aggregates of particles in the solution. By observing the molecules (dextran in our case) attached to the surface of the particles, the diameter determined by DLS measurements will be larger than that observed by TEM [
29]. According to previous studies [
30] it is possible that the particles may remain well dispersed/stable by coating with a polymer (in our case, dextran). It is known that when the particles are uncoated they tend to become agglomerated due to strong interparticle interactions.
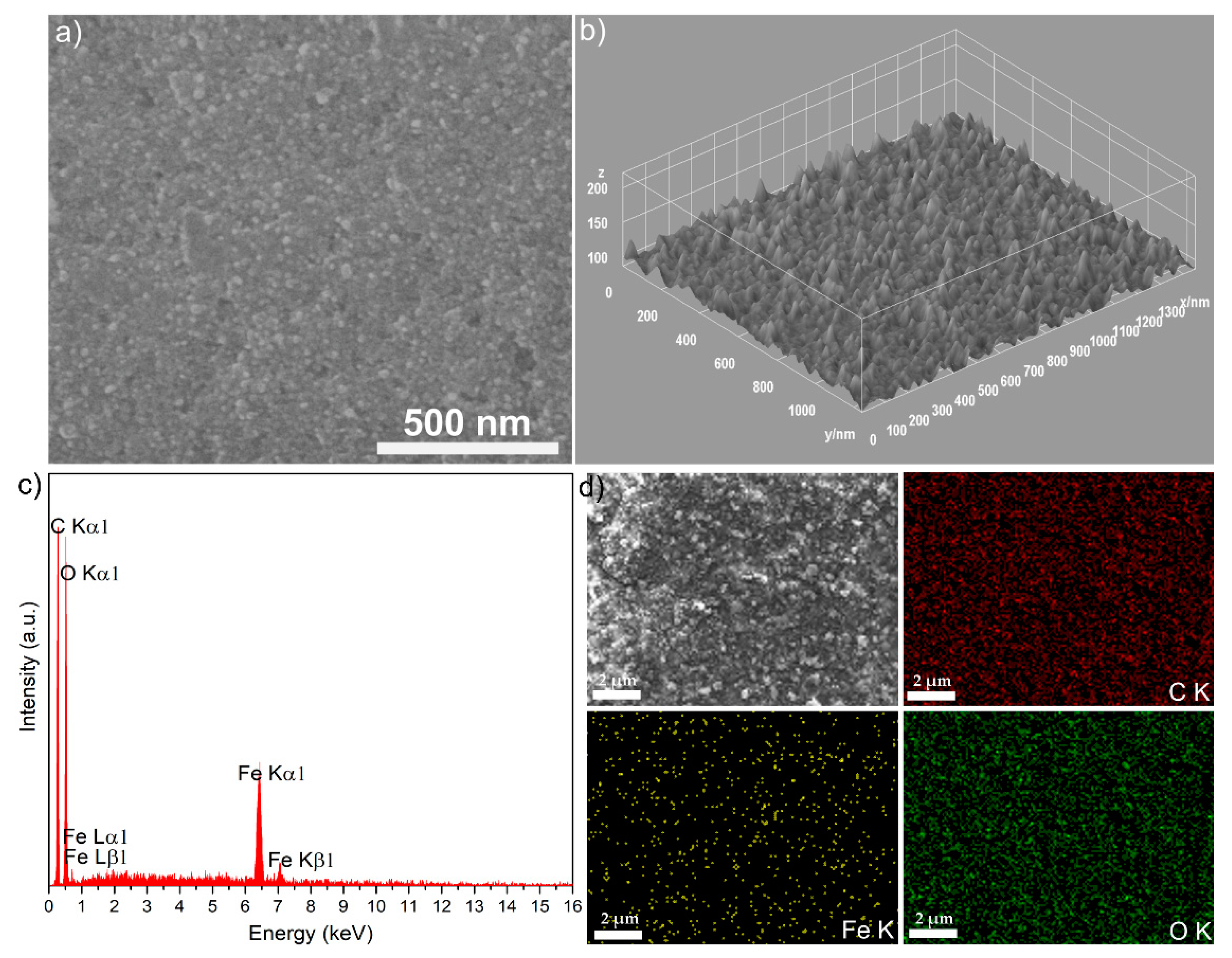
The morphology and thin surface layers obtained from the stable suspension of D-MNPs were evaluated by SEM investigations (
Figure 7a). Here, a SEM image of the surface D-MNP layer shows that the particles are spherical in shape. For a precise morphology assessment of the D-MNP layer, the 2D micrograph has been converted into a 3D surface map image (
Figure 7b) using specialized software [
31,
32,
33]. The chemical composition of the thin D-MNP layer was estimated by an Energy-dispersive X-ray spectroscopy (EDX) survey (
Figure 7c). The peaks of the typical chemical element constituents of the D-MNPs were identified (i.e., C, O, and Fe). No other peaks that could belong to impurities were observed. This result shows the good purity of the obtained D-MNPs. In the topographies of the elemental maps of the D-MNP layer, a homogeneous and uniform distribution of the constituent elements was revealed.
The toxicity of the D-MNP suspension and D-MNP layers were investigated using the first immortal human cell line (HeLa cells), which is known to be a remarkably durable and prolific cell line [
34]. The HeLa cells were incubated for 24 and 48 h with the D-MNP suspension and D-MNP layers, then visualized using a fluorescence microscope. The morphology of the HeLa cells treated with the D-MNP suspension and D-MNP layers after 24 and 48 h of incubation are presented in
Figure 8a–e.
Figure 8b,c depicts the morphology of the HeLa cells incubated for 24 h with the investigated samples. The morphology of the HeLa cells incubated for 48 h with the samples are depicted in
Figure 8e,f. The morphology of the HeLa cells grown in the culture medium for 24 and 48 h used as control are depicted in
Figure 8a,d. The results of the fluorescence microscopy revealed that the morphology of the HeLa cells incubated for 24 and 48 h with the D-MNP suspension and D-MNP layers did not change relative to the morphology presented by the control cells. Furthermore, the influence of the D-MNP suspension and D-MNP layers on the HeLa cell cycle was also investigated after 24 and 48 h of incubation by flow cytometry. The results of the cell cycle analysis are presented in
Figure 8g–l. The DNA histogram deconvolution was performed using the FlowJo software (FlowJo v9). The percentage of cells in the G0/G, S, and G2/M phases were quantified using the Watson model. It was observed that all tested compounds exhibited a small-scale level of cytotoxicity against HeLa cell cycle development for all of the tested time intervals. The flow cytometry histograms, depicted in
Figure 8g–l, are in good agreement with the microscopy images of the HeLa cell culture incubated for 24 and 48 h with D-MNP suspension and D-MNP layers. The cell cycle analysis demonstrated that both the D-MNP suspension and D-MNP layers did not present a significant toxicity towards HeLa cells, even after 48 h of incubation.
The cell cycle diagrams of the HeLa cells incubated for 24 and 48 h with the D-MNP suspension (
Figure 8h,k) and D-MNP layers (
Figure 8i,l) were comparable to the cell cycle diagrams obtained for the HeLa cells grown in culture medium used as control (
Figure 8g,j). The results of the percentage of cells in the G0/G, S, G2/M phases quantified using the Watson model revealed that there was no blockage of the eukaryotic G2/M phase, indicating no toxicity.
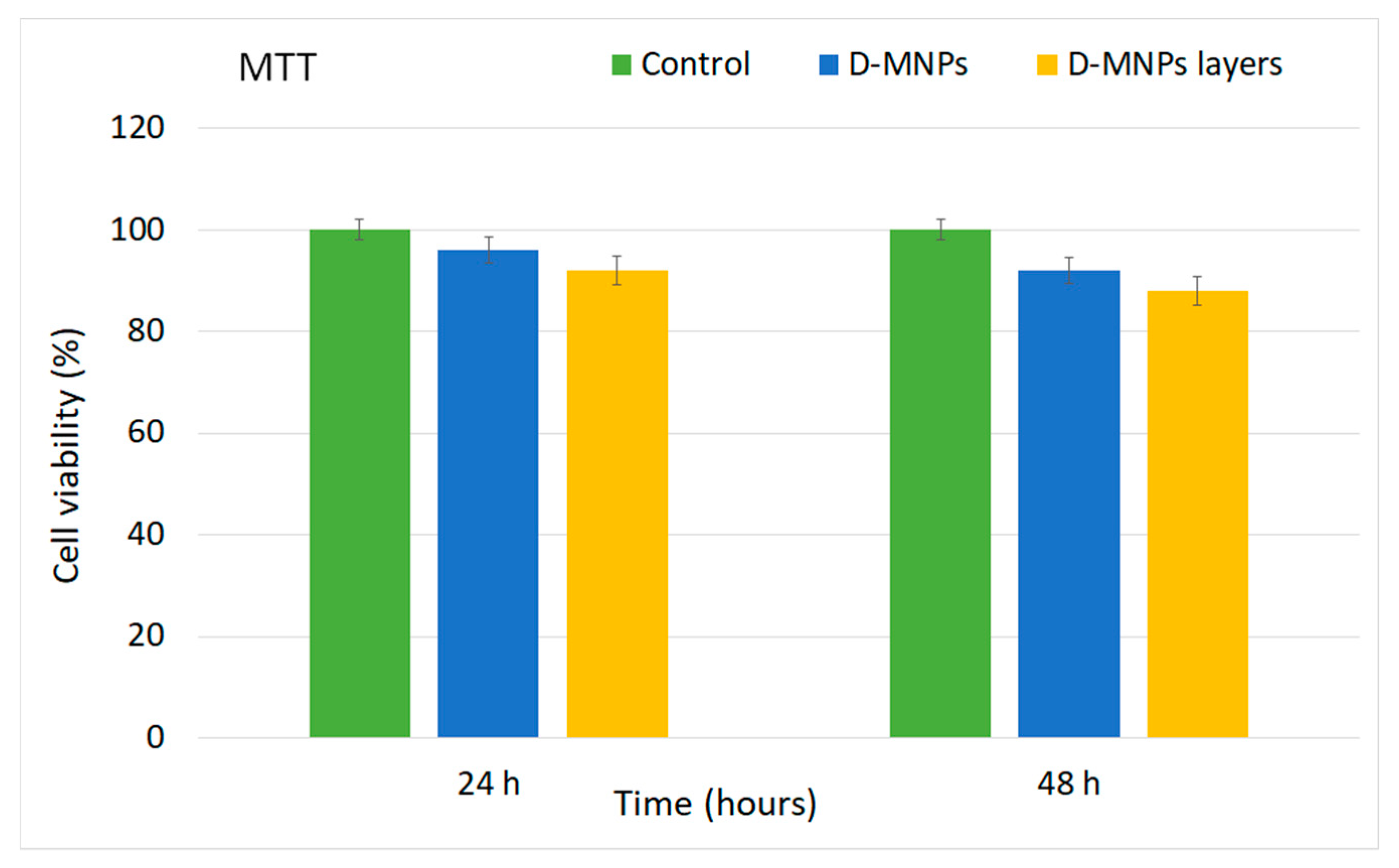
In order to assess the cytotoxicity of the D-MNP suspension and D-MNP layers, a quantitative MTT assay was performed to determine the cell viability of the HeLa cells after the 24 and 48 h incubation time intervals, in the presence of the D-MNP suspension and D-MNP layers. The results of the cytotoxicity tests at different incubation time periods (24 and 48 h) of the D-MNP suspension and D-MNP layers on the HeLa cell lines are shown in
Figure 9. The MTT studies revealed that after 24 h of incubation time, there were no representative differences in the values of cells viabilities between the cells treated with D-MNPs suspension and D-MNPs layers and the control cell culture. The results have emphasized that after 24 h of incubation, the cell viability of HeLa cells was 96% in the case of the D-MNP suspension and 92% in the case of the D-MNP layers. Moreover, the MTT assay results show that after 48 h of incubation a slight decrease in HeLa cell viability was observed for both samples. In this case, a cell viability of 92% was obtained in the case of HeLa cells exposed to the D-MNP suspension and a cell viability of 88% was obtained in the case of HeLa cells exposed to the D-MNP layers. The results highlight that there is a correlation between the incubation time and the small toxicity effects presented by the tested samples. Moreover, the MTT suggested that the D-MNP suspension had better biocompatible properties than the D-MNP layers. The results obtained in the present study are in good agreement with previous studies regarding the toxicity of iron oxide nanoparticles [
35,
36,
37,
38,
39,
40,
41,
42]. Moreover, the results obtained from the statistical analysis, where
p = 0.042, demonstrated that the observed difference observed in the cytotoxicity assays is unlikely to be due to chance, indicating a significant finding.
Usually, the cytotoxicity induced by nanomaterials and nanolayers depends on numerous parameters, such as the type of the nanoparticles, their size and shape, the concentration, incubation time, and also the type of cell line [
35,
36,
37,
38]. In their study regarding, the “synthesis, characterization and toxicological evaluation of iron oxide nanoparticles in human lung alveolar epithelial cells”, Dwivedi et al. [
39] reported that the cell viability of A-549 cells diminished to 84%, 72%, and 56% after being incubated for 24 h with different concentrations, specifically, 10, 25, and 50 μg/mL, respectively, of iron oxide nanoparticles (IONPs). Another study conducted by Karlsson et al. [
40] regarding the cytotoxicity of several metal oxide nanoparticles reported that iron oxide particles (Fe
3O
4, Fe
2O
3) exhibited none or low toxicity, while CuZnFe
2O
4 particles induced DNA damage to the A549 cell line. During recent years, several materials have been employed as coatings for IONPs in order to stabilize their physicochemical and biological properties. Villanueva et al. [
41] studied “the influence of surface functionalization on the enhanced internalization of magnetic nanoparticles in cancer cells”, and their results emphasize that iron oxide nanoparticles coated with dextranamino at concentrations of 0.05, 0.1, and 0.5 mg/mL present a low toxicity against HeLa cells. Moreover, Ankamwar et al. [
42] demonstrated in their study that IONPs coated with a bipolar surfactant, namely, tetramethylammonium 11-aminoundecanoate, at concentrations of 0.1 to 10 μg/mL had no toxicity against HeLa cells. Furthermore, they have reported that the cytotoxicity of IONPs coated with a bipolar surfactant, tetramethylammonium 11-aminoundecanoate, was strongly dependent on the nanoparticle concentration [
42].



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}