
Functional Interplay between P5 and PDI/ERp72 to Drive Protein Folding

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Plasmid Construction
2.3. Recombinant Protein Expression and Purification
2.4. Far-Western Dot Blotting
2.5. Isothermal Titration Calorimetry
2.6. Preparation of Reduced and Denatured RNase A
2.7. Gel-Shift Assay of RNase A Oxidation
2.8. RNase A Reactivation Assay
2.9. GAPDH Aggregation Assay
2.10. Statistical Analysis
3. Results
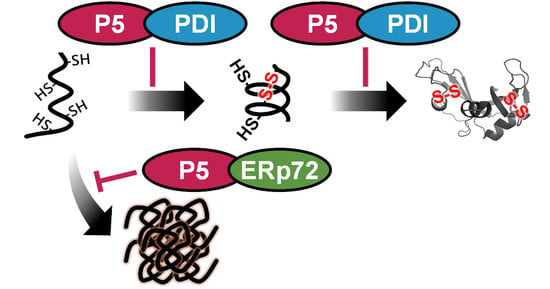
3.1. PDI and ERp72 Form Non-Covalent Complexes with P5
3.2. P5 and PDI Act in Concert to Synergistically Accelerate Oxidative Folding
3.3. P5 Acts in Concert with ERp72 to Strongly Inhibit Client Aggregation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arolas, J.L.; Aviles, F.X.; Chang, J.-Y.; Ventura, S. Folding of small disulfide-rich proteins: Clarifying the puzzle. Trends Biochem. Sci. 2006, 31, 292–301. [Google Scholar] [CrossRef] [Green Version]
- Narayan, M. Revisiting the Formation of a Native Disulfide Bond: Consequences for Protein Regeneration and Beyond. Molecules 2020, 25, 5337. [Google Scholar] [CrossRef] [PubMed]
- Narayan, M. The Structure-Forming Juncture in Oxidative Protein Folding: What happens in the ER? Adv. Exp. Med. Biol. 2017, 966, 163. [Google Scholar] [CrossRef] [Green Version]
- Kopito, R.R.; Sitia, R. Aggresomes and Russell bodies: Symptoms of cellular indigestion? EMBO Rep. 2000, 1, 225. [Google Scholar] [CrossRef] [Green Version]
- Ushioda, R.; Hoseki, J.; Araki, K.; Jansen, G.; Thomas, D.Y.; Nagata, K. ERdj5 Is Required as a Disulfide Reductase for Degradation of Misfolded Proteins in the ER. Science 2008, 321, 569–572. [Google Scholar] [CrossRef]
- Fraga, H.; Graña-Montes, R.; Illa, R.; Covaleda, G.; Ventura, S. Association Between Foldability and Aggregation Propensity in Small Disulfide-Rich Proteins. Antioxid. Redox Signal. 2014, 21, 368–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcduffee, A.T.; Senisterra, G.; Huntley, S.; Lepock, J.R.; Sekhar, K.R.; Meredith, M.J.; Borrelli, M.J.; Morrow, J.D.; Freeman, M.L. Proteins Containing Non-Native Disulfide Bonds Generated by Oxidative Stress Can Act as Signals for the Induction of the Heat Shock Response. J. Cell. Physiol. 1997, 171, 143–151. [Google Scholar] [CrossRef]
- Meyer, A.J.; Riemer, J.; Rouhier, N. Oxidative protein folding: State-of-the-art and current avenues of research in plants. New Phytol. 2019, 221, 1230–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Inaba, K. Disulfide bond formation network in the three biological kingdoms, bacteria, fungi and mammals. FEBS J. 2012, 279, 2262–2271. [Google Scholar] [CrossRef]
- Urade, R. Oxidative protein folding in the plant endoplasmic reticulum. Biosci. Biotechnol. Biochem. 2019, 83, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, S.; Matsusaki, M.; Inaba, K.; Okumura, M. PDI family members as guides for client folding and assembly. Int. J. Mol. Sci. 2020, 21, 9351. [Google Scholar] [CrossRef]
- Kojima, R.; Okumura, M.; Masui, S.; Kanemura, S.; Inoue, M.; Saiki, M.; Yamaguchi, H.; Hikima, T.; Suzuki, M.; Akiyama, S.; et al. Radically different thioredoxin domain arrangement of ERp46, an efficient disulfide bond introducer of the mammalian PDI family. Structure 2014, 22, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Kojima, R.; Okumura, M.; Hagiwara, M.; Masui, S.; Maegawa, K.; Saiki, M.; Horibe, T.; Suzuki, M.; Inaba, K. Synergistic cooperation of PDI family members in peroxiredoxin 4-driven oxidative protein folding. Sci. Rep. 2013, 3, 2456. [Google Scholar] [CrossRef]
- Okumura, M.; Noi, K.; Inaba, K. Visualization of structural dynamics of protein disulfide isomerase enzymes in catalysis of oxidative folding and reductive unfolding. Curr. Opin. Struct. Biol. 2021, 66, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Matsusaki, M.; Kanemura, S.; Kinoshita, M.; Lee, Y.-H.; Inaba, K.; Okumura, M. the protein disulfide isomerase family: From proteostasis to pathogenesis. Biochim. Biophys. Acta-Gen. Subj. 2020, 1864, 129338. [Google Scholar] [CrossRef]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.-Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Woehlbier, U.; Colombo, A.; Saaranen, M.J.; Pérez, V.; Ojeda, J.; Bustos, F.J.; Andreu, C.I.; Torres, M.; Valenzuela, V.; Medinas, D.B.; et al. ALS-linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 2016, 35, 845–865. [Google Scholar] [CrossRef]
- Walker, A.K.; Farg, M.A.; Bye, C.R.; McLean, C.A.; Horne, M.K.; Atkin, J.D. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 2010, 133, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-B.; Shi, Q.; Xu, Y.; Xie, W.-L.; Zhang, J.; Tian, C.; Guo, Y.; Wang, K.; Zhang, B.-Y.; Chen, C.; et al. Protein disulfide isomerase regulates endoplasmic reticulum stress and the apoptotic process during prion infection and PrP mutant-induced cytotoxicity. PLoS ONE 2012, 7, e38221. [Google Scholar] [CrossRef]
- Yu, J.; Li, T.; Liu, Y.; Wang, X.; Zhang, J.; Wang, X.; Shi, G.; Lou, J.; Wang, L.; Wang, C.; et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020, 39, e103841. [Google Scholar] [CrossRef]
- Wang, C.; Yu, J.; Huo, L.; Wang, L.; Feng, W.; Wang, C. Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a’. J. Biol. Chem. 2012, 287, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Wang, C.C.; Tsou, C.L. Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J. Biol. Chem. 1994, 269, 24550–24552. [Google Scholar] [CrossRef]
- Okumura, M.; Kadokura, H.; Hashimoto, S.; Yutani, K.; Kanemura, S.; Hikima, T.; Hidaka, Y.; Ito, L.; Shiba, K.; Masui, S.; et al. Inhibition of the functional interplay between endoplasmic reticulum (ER) oxidoreduclin-1α (Ero1α) and protein-disulfide isomerase (PDI) by the endocrine disruptor bisphenol A. J. Biol. Chem. 2014, 289, 27004–27018. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Wang, C. Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese. Eur. J. Biochem. 1995, 231, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Wang, C. Both the isomerase and chaperone activities of protein disulfide isomerase are required for the reactivation of reduced and denatured acidic phospholipase A2. EMBO J. 1997, 16, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Klappa, P.; Freedman, R.B.; Lilie, H.; Rudolph, R. Catalytic activity and chaperone function of human protein-disulfide isomerase are required for the efficient refolding of proinsulin. J. Biol. Chem. 2002, 277, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puig, A.; Gilbert, H.F. Protein disulfide isomerase exhibits chaperone and anti-chaperone activity in the oxidative refolding of lysozyme. J. Biol. Chem. 1994, 269, 7764–7771. [Google Scholar] [CrossRef]
- Quan, H.; Fan, G.; Wang, C.C. Independence of the chaperone activity of protein disulfide isomerase from its thioredoxin-like active site. J. Biol. Chem. 1995, 270, 17078–17080. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.X.; Dai, Y.; Liu, H.P.; Chen, S.M.; Wang, C. chen Contributions of protein disulfide isomerase domains to its chaperone activity. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2000, 1481, 45–54. [Google Scholar] [CrossRef]
- Parakh, S.; Atkin, J.D. Novel roles for protein disulphide isomerase in disease states: A double edged sword? Front. Cell Dev. Biol. 2015, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Okumura, M.; Noi, K.; Kanemura, S.; Kinoshita, M.; Saio, T.; Inoue, Y.; Hikima, T.; Akiyama, S.; Ogura, T.; Inaba, K. Dynamic assembly of protein disulfide isomerase in catalysis of oxidative folding. Nat. Chem. Biol. 2019, 15, 499–509. [Google Scholar] [CrossRef]
- Gjaltema, R.A.F.; Bank, R.A. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106. [Google Scholar] [CrossRef] [PubMed]
- Wetterau, J.R.; Lin, M.C.M.; Jamil, H. Microsomal triglyceride transfer protein. Biochim. Biophys. Acta-Lipids Lipid Metab. 1997, 1345, 136–150. [Google Scholar] [CrossRef] [Green Version]
- Okumura, M.; Kanemura, S.; Matsusaki, M.; Kinoshita, M.; Saio, T.; Ito, D.; Hirayama, C.; Kumeta, H.; Watabe, M.; Amagai, Y.; et al. A unique leucine-valine adhesive motif supports structure and function of protein disulfide isomerase P5 via dimerization. Structure 2021. [Google Scholar] [CrossRef]
- Kikuchi, M.; Doi, E.; Tsujimoto, I.; Horibe, T.; Tsujimoto, Y. Functional analysis of human P5, a protein disulfide isomerase homologue. J. Biochem. 2002, 132, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuy, A.; Passam, F. Functional assays of thiol isomerase ERp5. Methods Mol. Biol. 2019, 1967, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Eletto, D.D.; Eletto, D.D.; Dersh, D.; Gidalevitz, T.; Argon, Y. protein disulfide isomerase a6 controls the decay of IRE1α Signaling via disulfide-dependent association. Mol. Cell 2014, 53, 562–576. [Google Scholar] [CrossRef] [Green Version]
- Eletto, D.; Chevet, E.; Argon, Y.; Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 2014, 127, 3649–3658. [Google Scholar] [CrossRef] [Green Version]
- Groenendyk, J.; Peng, Z.; Dudek, E.; Fan, X.; Mizianty, M.J.; Dufey, E.; Urra, H.; Sepulveda, D.; Rojas-Rivera, D.; Lim, Y.; et al. Interplay between the oxidoreductase PDIA6 and microRNA-322 Controls the response to disrupted endoplasmic reticulum calcium homeostasis. Sci. Signal. 2014, 7, ra54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Iemura, S.I.; Kamiya, Y.; Ron, D.; Kato, K.; Natsume, T.; Nagata, K. Ero1-α and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 2013, 202, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Kanemura, S.; Okumura, M.; Yutani, K.; Ramming, T.; Hikima, T.; Appenzeller-Herzog, C.; Akiyama, S.; Inaba, K. Human ER Oxidoreductin-1α (Ero1α) undergoes dual regulation through complementary redox interactions with protein-disulfide isomerase. J. Biol. Chem. 2016, 291, 23952–23964. [Google Scholar] [CrossRef] [Green Version]
- Matsusaki, M.; Okuda, A.; Masuda, T.; Koishihara, K.; Mita, R.; Iwasaki, K.; Hara, K.; Naruo, Y.; Hirose, A.; Tsuchi, Y.; et al. Cooperative protein folding by two protein thiol disulfide oxidoreductases and ERO1 in soybean. Plant Physiol. 2016, 170, 774–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusaki, M.; Okuda, A.; Matsuo, K.; Gekko, K.; Masuda, T.; Naruo, Y.; Hirose, A.; Kono, K.; Tsuchi, Y.; Urade, R. Regulation of plant ER oxidoreductin 1 (ERO1) activity for efficient oxidative protein folding. J. Biol. Chem. 2019, 294, 18820–18835. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Kinoshita, M.; Kume, S.; GT, H.; Sugiki, T.; Ladbury, J.E.; Kojima, C.; Ikegami, T.; Kurisu, G.; Goto, Y.; et al. Non-covalent forces tune the electron transfer complex between ferredoxin and sulfite reductase to optimize enzymatic activity. Biochem. J. 2016, 473, 3837–3854. [Google Scholar] [CrossRef] [Green Version]
- Creighton, T.E. Kinetics of refolding of reduced ribonuclease. J. Mol. Biol. 1977, 113, 329–341. [Google Scholar] [CrossRef]
- Wang, L.; Li, S.; Sidhu, A.; Zhu, L.; Liang, Y.; Freedman, R.B.; Wang, C. Reconstitution of human Ero1-Lalpha/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a’ domains of protein-disulfide isomerase. J. Biol. Chem. 2009, 284, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, A.-M.; Besir, H. Staining of proteins in gels with coomassie G-250 without organic solvent and acetic acid. J. Vis. Exp. 2009. [Google Scholar] [CrossRef] [PubMed]
- Lyles, M.M.; Gilbert, H.F. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: Dependence of the rate on the composition of the redox buffer. Biochemistry 1991, 30, 613–619. [Google Scholar] [CrossRef]
- Saio, T.; Guan, X.; Rossi, P.; Economou, A.; Kalodimos, C.G. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 2014, 344, 1250494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.-C.; Li, Z.-Y.; Zhou, J.-M.; Fischer, G. Assisted folding of D-glyceraldehyde-3-phosphate dehydrogenase by trigger factor. Protein Sci. 2000, 9, 1254–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, G.; Patzelt, H.; Rauch, T.; Kurz, T.A.; Vorderwülbecke, S.; Bukau, B.; Deuerling, E. Trigger factor peptidyl-prolyl cis/trans isomerase activity is not essential for the folding of cytosolic proteins in escherichia coli. J. Biol. Chem. 2004, 279, 14165–14170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Li, Q.; Chen, X.-Z. Detecting protein–protein interactions by far western blotting. Nat. Protoc. 2007, 2, 3278–3284. [Google Scholar] [CrossRef] [PubMed]
- Walsh, B.W.; Lenhart, J.S.; Schroeder, J.W.; Simmons, L.A. Far western blotting as a rapid and efficient method for detecting interactions between DNA replication and DNA repair proteins. In Single-Stranded DNA Binding Proteins; Humana Press: Totowa, NJ, USA, 2012; pp. 161–168. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, T.; Suno, R.; Iemura, S.I.; Natsume, T.; Wada, I.; Hosokawa, N. Endoplasmic reticulum proteins SDF2 and SDF2L1 act as components of the BiP chaperone cycle to prevent protein aggregation. Genes Cells 2017, 22, 684–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanikawa, Y.; Kanemura, S.; Ito, D.; Lin, Y.; Matsusaki, M.; Kuroki, K.; Yamaguchi, H.; Maenaka, K.; Lee, Y.-H.; Inaba, K.; et al. Ca2+ regulates ERp57-calnexin complex formation. Molecules 2021, 26, 2853. [Google Scholar] [CrossRef]
- Zhao, R. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the Hsp90 Chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef] [Green Version]
- Lepvrier, E.; Nigen, M.; Moullintraffort, L.; Chat, S.; Allegro, D.; Barbier, P.; Thomas, D.; Nazabal, A.; Garnier, C. Hsp90 oligomerization process: How can p23 drive the chaperone machineries? Biochim. Biophys. Acta-Proteins Proteom. 2015, 1854, 1412–1424. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Grantham, J. The molecular chaperone CCT/TRiC: An essential component of proteostasis and a potential modulator of protein aggregation. Front. Genet. 2020, 0, 172. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S. Molecular chaperones: From proteostasis to pathogenesis. FEBS J. 2018, 285, 3353–3361. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsusaki, M.; Okada, R.; Tanikawa, Y.; Kanemura, S.; Ito, D.; Lin, Y.; Watabe, M.; Yamaguchi, H.; Saio, T.; Lee, Y.-H.; et al. Functional Interplay between P5 and PDI/ERp72 to Drive Protein Folding. Biology 2021, 10, 1112. https://doi.org/10.3390/biology10111112
Matsusaki M, Okada R, Tanikawa Y, Kanemura S, Ito D, Lin Y, Watabe M, Yamaguchi H, Saio T, Lee Y-H, et al. Functional Interplay between P5 and PDI/ERp72 to Drive Protein Folding. Biology. 2021; 10(11):1112. https://doi.org/10.3390/biology10111112
Chicago/Turabian StyleMatsusaki, Motonori, Rina Okada, Yuya Tanikawa, Shingo Kanemura, Dai Ito, Yuxi Lin, Mai Watabe, Hiroshi Yamaguchi, Tomohide Saio, Young-Ho Lee, and et al. 2021. "Functional Interplay between P5 and PDI/ERp72 to Drive Protein Folding" Biology 10, no. 11: 1112. https://doi.org/10.3390/biology10111112
APA StyleMatsusaki, M., Okada, R., Tanikawa, Y., Kanemura, S., Ito, D., Lin, Y., Watabe, M., Yamaguchi, H., Saio, T., Lee, Y.-H., Inaba, K., & Okumura, M. (2021). Functional Interplay between P5 and PDI/ERp72 to Drive Protein Folding. Biology, 10(11), 1112. https://doi.org/10.3390/biology10111112