
A Novel WT1 Mutation Identified in a 46,XX Testicular/Ovotesticular DSD Patient Results in the Retention of Intron 9

 , , , , , ,
, , , , , ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. 46,XX TDSD/OTDSD Case Report
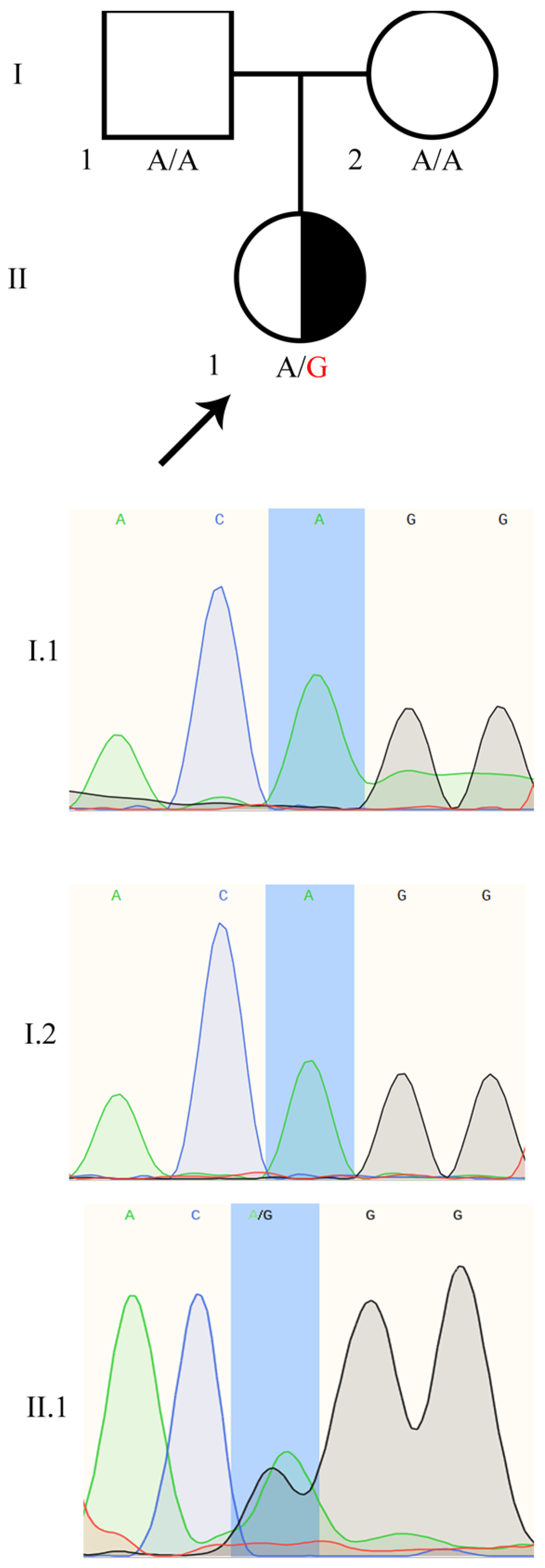
2.2. Identification of the Heterozygous WT1:c.1437A>G Mutation in the Patient
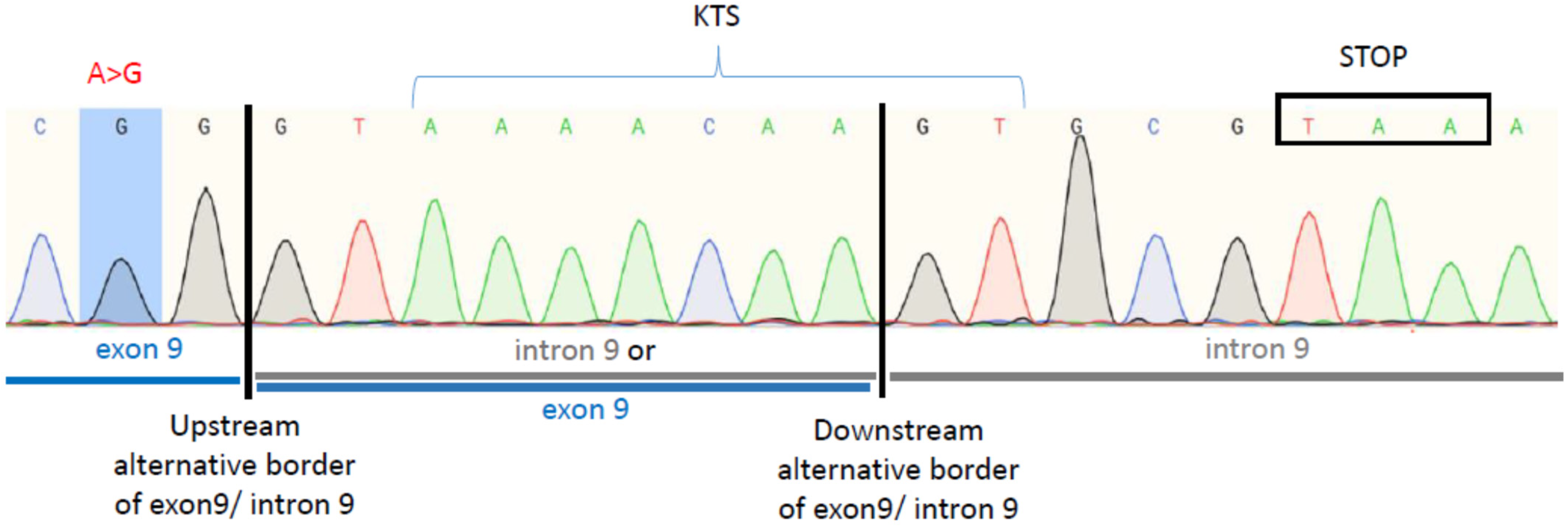
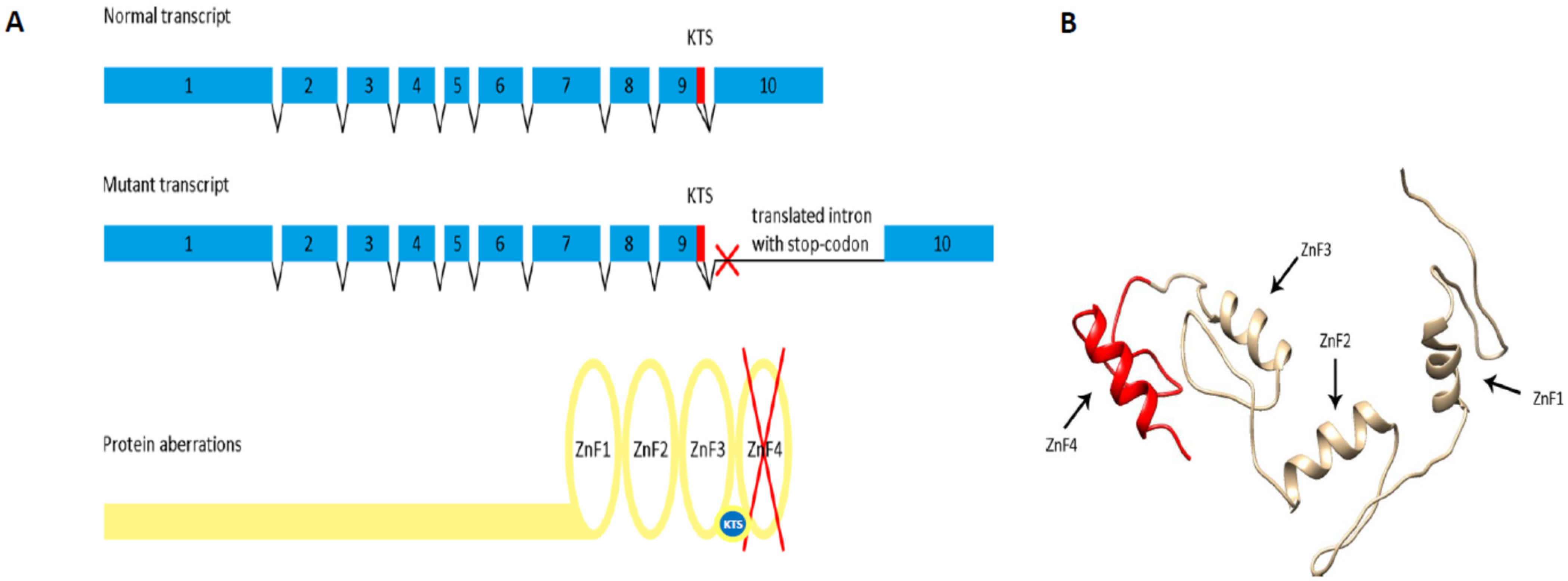
2.3. The WT1:c.1437A>G Mutation Results in the Retention of Intron 9, Predicting Premature Translational Termination and Absence of Zinc Finger 4
3. Discussion
4. Materials and Methods
4.1. Patient Description
4.2. Hormonal Analysis
4.3. Cytogenetic Studies
4.4. Whole Exome Sequencing (WES)
4.5. The WT1:c.1437A>G Variant Validation by Sanger Sequencing
4.6. Bioinformatic Resources
4.7. Influence of the WT1:c.1437A>G Variant on Splicing
4.8. D Protein Modelling
4.9. Accession Numbers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eggers, S.; Ohnesorg, T.; Sinclair, A. Genetic regulation of mammalian gonad development. Nat. Rev. Endocrinol. 2014, 10, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Lamothe, S.; Bernard, V.; Christin-Maitre, S. Gonad differentiation toward ovary. Ann. d’Endocrinol. 2020, 81, 83–88. [Google Scholar] [CrossRef]
- Lee, P.A.; Nordenstrom, A.; Houk, C.P.; Ahmed, S.F.; Auchus, R.; Baratz, A.; Baratz Dalke, K.; Liao, L.M.; Lin-Su, K.; Looijenga, L.H., 3rd; et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr. 2016, 85, 158–180. [Google Scholar] [CrossRef]
- De La Chapelle, A. Analytic review: Nature and origin of males with XX sex chromosomes. Am. J. Hum. Genet. 1972, 24, 71–105. [Google Scholar]
- Kilberg, M.J.; McLoughlin, M.; Pyle, L.C.; Vogiatzi, M.G. Endocrine Management of Ovotesticular DSD, an Index Case and Review of the Literature. Pediatr. Endocrinol. Rev. 2019, 17, 110–116. [Google Scholar] [PubMed]
- Croft, B.; Ohnesorg, T.; Hewitt, J.; Bowles, J.; Quinn, A.; Tan, J.; Corbin, V.; Pelosi, E.; Bergen, J.V.D.; Sreenivasan, R.; et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat. Commun. 2018, 9, 5319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyon, C.; Chantot-Bastaraud, S.; Harbuz, R.; Bhouri, R.; Perrot, N.; Peycelon, M.; Sibony, M.; Rojo, S.; Piguel, X.; Bilan, F.; et al. Refining the regulatory region upstream ofSOX9associated with 46,XX testicular disorders of Sex Development (DSD). Am. J. Med. Genet. Part A 2015, 167, 1851–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeherunvong, T.; Perera, E.M.; Bao, Y.; Benke, P.J.; Benigno, A.; Donahue, R.P.; Berkovitz, G.D. 46,XX sex reversal with partial duplication of chromosome arm 22q. Am. J. Med. Genet. 2004, 127A, 149–151. [Google Scholar] [CrossRef]
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J. Clin. Investig. 2011, 121, 328–341. [Google Scholar] [CrossRef] [Green Version]
- Biason-Lauber, A.; Konrad, D.; Navratil, F.; Schoenle, E.J. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N. Engl. J. Med. 2004, 351, 792–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parma, P.; Radi, O.; Vidal, V.; Chaboissier, M.-C.; Dellambra, E.; Valentini, S.; Guerra, L.; Schedl, A.; Camerino, G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 2006, 38, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Eozenou, C.; Gonen, N.; Touzon, M.S.; Jorgensen, A.; Yatsenko, S.A.; Fusee, L.; Kamel, A.K.; Gellen, B.; Guercio, G.; Singh, P.; et al. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms’ tumor 1 (WT1) gene. Proc. Natl. Acad. Sci. USA 2020, 117, 13680–13688. [Google Scholar] [CrossRef]
- Gomes, N.L.; de Paula, L.C.; Silva, J.M.; Silva, T.E.; Lerário, A.M.; Nishi, M.Y.; Batista, R.L.; Júnior, J.A.D.F.; Moraes, D.; Costa, E.M.; et al. A 46,XX testicular disorder of sex development caused by a Wilms’ tumour Factor-1 (WT1) pathogenic variant. Clin. Genet. 2019, 95, 172–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef]
- Kent, J.; Coriat, A.M.; Sharpe, P.; Hastie, N.D.; van Heyningen, V. The evolution of WT1 sequence and expression pattern in the vertebrates. Oncogene 1995, 11, 1781–1792. [Google Scholar]
- Laity, J.H.; Chung, J.; Dyson, H.J.; Wright, P.E. Alternative Splicing of Wilms’ Tumor Suppressor Protein Modulates DNA Binding Activity through Isoform-Specific DNA-Induced Conformational Changes. Biochemistry 2000, 39, 5341–5348. [Google Scholar] [CrossRef]
- Ullmark, T.; Montano, G.; Gullberg, U. DNA and RNA binding by the Wilms’ tumour gene 1 (WT1) protein +KTS and −KTS isoforms-from initial observations to recent global genomic analyses. Eur. J. Haematol. 2018, 100, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Bharathavikru, R.; Dudnakova, T.; Aitken, S.; Slight, J.; Artibani, M.; Hohenstein, P.; Tollervey, D.; Hastie, N. Transcription factor Wilms’ tumor 1 regulates developmental RNAs through 3′ UTR interaction. Genes Dev. 2017, 31, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Davies, R.C.; Calvio, C.; Bratt, E.; Larsson, S.H.; Lamond, A.I.; Hastie, N.D. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes Dev. 1998, 12, 3217–3225. [Google Scholar] [CrossRef] [Green Version]
- Ullmark, T.; Järvstråt, L.; Sandén, C.; Montano, G.; Jernmark-Nilsson, H.; Lilljebjörn, H.; Lennartsson, A.; Fioretos, T.; Drott, K.; Vidovic, K.; et al. Distinct global binding patterns of the Wilms tumor gene 1 (WT1) −KTS and +KTS isoforms in leukemic cells. Haematologica 2016, 102, 336–345. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.Y.; Qiu, Q.Q.; Deuel, T.F. The Wilms’ tumor gene product WT1 activates or suppresses transcription through separate functional domains. J. Biol. Chem. 1993, 268, 9172–9175. [Google Scholar] [CrossRef]
- Reddy, J.C.; Morris, J.C.; Wang, J.; English, M.A.; Haber, D.A.; Shi, Y.; Licht, J.D. WT1-mediated Transcriptional Activation Is Inhibited by Dominant Negative Mutant Proteins. J. Biol. Chem. 1995, 270, 10878–10884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammes, A.; Guo, J.-K.; Lutsch, G.; Leheste, J.-R.; Landrock, D.; Ziegler, U.; Gubler, M.-C.; Schedl, A. Two Splice Variants of the Wilms’ Tumor 1 Gene Have Distinct Functions during Sex Determination and Nephron Formation. Cell 2001, 106, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Barbaux, S.; Niaudet, P.; Gubler, M.-C.; Grünfeld, J.-P.; Jaubert, F.; Kuttenn, F.; Fékété, C.N.; Souleyreau-Therville, N.; Thibaud, E.; Fellous, M.; et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat. Genet. 1997, 17, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Klamt, B.; Koziell, A.; Poulat, F.; Wieacker, P.; Scambler, P.; Berta, P.; Gessler, M. Frasier syndrome is caused by defective alternative splicing of WT1 leading to an altered ratio of WT1 +/−KTS splice isoforms. Hum. Mol. Genet. 1998, 7, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Audí, L.; Ahmed, S.F.; Krone, N.; Cools, M.; McElreavey, K.; Holterhus, P.-M.; Greenfield, A.; Bashamboo, A.; Hiort, O.; Wudy, S.A.; et al. GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): Position paper of EU COST Action BM 1303 ‘DSDnet’. Eur. J. Endocrinol. 2018, 179, R197–R206. [Google Scholar] [CrossRef] [Green Version]
- Baxter, R.M.; Arboleda, V.; Lee, H.; Barseghyan, H.; Adam, M.P.; Fechner, P.Y.; Bargman, R.; Keegan, C.; Travers, S.; Schelley, S.; et al. Exome Sequencing for the Diagnosis of 46,XY Disorders of Sex Development. J. Clin. Endocrinol. Metab. 2015, 100, E333–E344. [Google Scholar] [CrossRef]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Zhang, X.; Wang, L.; Wang, R.; Huang, Z.; Sun, Y.; Yao, R.; Huang, X.; Ye, J.; Han, L.; et al. Diagnostic Application of Targeted Next-Generation Sequencing of 80 Genes Associated with Disorders of Sexual Development. Sci. Rep. 2017, 7, 44536. [Google Scholar] [CrossRef] [Green Version]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, R.H.; Du, K.; Lee, V.M.; Mohn, K.L.; Haber, B.A.; Tewari, D.S.; Taub, R. Novel delayed-early and highly insulin-induced growth response genes. Identification of HRS, a potential regulator of alternative pre-mRNA splicing. J. Biol. Chem. 1993, 268, 15185–15192. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.; Bruening, W.; Kashtan, C.E.; Mauer, S.M.; Manivel, J.C.; Striegel, J.E.; Houghton, D.C.; Junien, C.; Habib, R.; Fouser, L. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 1991, 67, 437–447. [Google Scholar] [CrossRef]
- Mueller, R.F. The Denys-Drash syndrome. J. Med. Genet. 1994, 31, 471–477. [Google Scholar] [CrossRef] [Green Version]
- Nurmemmedov, E.; Yengo, R.K.; Uysal, H.; Karlsson, R.; Thunnissen, M.M. New insights into DNA-binding behavior of Wilms Tumor Protein (WT1)—A dual study. Biophys. Chem. 2009, 145, 116–125. [Google Scholar] [CrossRef]
- Stoll, R.; Lee, B.M.; Debler, E.W.; Laity, J.H.; Wilson, I.A.; Dyson, H.J.; Wright, P.E. Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol. 2007, 372, 1227–1245. [Google Scholar] [CrossRef]
- Bardeesy, N.; Pelletier, J. Overlapping RNA and DNA binding domains of the WT1 tumor suppressor gene product. Nucleic Acids Res. 1998, 26, 1784–1792. [Google Scholar] [CrossRef] [Green Version]
- Chassot, A.A.; Ranc, F.; Gregoire, E.P.; Roepers-Gajadien, H.L.; Taketo, M.M.; Camerino, G.; de Rooij, D.G.; Schedl, A.; Chaboissier, M.C. Activation of beta-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum. Mol. Genet. 2008, 17, 1264–1277. [Google Scholar] [CrossRef] [Green Version]
- Fagerlund, R.D.; Ooi, P.L.; Wilbanks, S.M. Soluble expression and purification of tumor suppressor WT1 and its zinc finger domain. Protein Expr. Purif. 2012, 85, 165–172. [Google Scholar] [CrossRef]
- Moffett, P.; Bruening, W.; Nakagama, H.; Bardeesy, N.; Housman, D.E.; Pelletier, J. Antagonism of WT1 activity by protein self-association. Proc. Natl. Acad. Sci. USA 1995, 92, 11105–11109. [Google Scholar] [CrossRef] [Green Version]
- Bertelloni, S.; Russo, G.; Baroncelli, G.I. Human Chorionic Gonadotropin Test: Old Uncertainties, New Perspectives, and Value in 46,XY Disorders of Sex Development. Sex. Dev. 2018, 12, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed] [Green Version]
- Karczewski, K.J.; Weisburd, B.; Thomas, B.; Solomonson, M.; Ruderfer, D.M.; Kavanagh, D.; Hamamsy, T.; Lek, M.; Samocha, K.; Cummings, B.; et al. The ExAC browser: Displaying reference data information from over 60,000 exomes. Nucleic Acids Res. 2017, 45, D840–D845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hormone, Unit | Age 2 Months | Age 2 Months Reference Values | Age 14 Months | Age 14 Months Reference Values | |
|---|---|---|---|---|---|
| fT, ng/dL | Baseline | 0.001 | ♀ < 0.5 ♂ < 0.5 | ||
| 24 h after 3-days hCG stimulation | 0.321 | ||||
| DHT, ng/dL | 1.186 | ♀ < 3 ♂ < 3 | |||
| AMH, ng/mL | 10.56 | ♀ 0.17–8.9 ♂ 3.8–159.8 | |||
| E2, pg/mL | 0.23 | ♀ < 15 ♂ < 15 | |||
| LH, mIU/mL | 11.19 | ♀ 0.02–7.0 ♂ 0.02–7.0 | <0.1 | ♀ 0.02–0.3 ♂ 0.02–0.3 | |
| FSH, mIU/mL | 6.43 | ♀ 0.24–14.2 ♂ 0.16–4.1 | 1.01 | ♀ 0.3–11.1 ♂1.0–4.2 | |
| 17-OHP, ng/mL | 0.43 | ♀ 0.1–4.0 ♂ 0.1–4.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirokha, D.; Gorodna, O.; Vitrenko, Y.; Zelinska, N.; Ploski, R.; Nef, S.; Jaruzelska, J.; Kusz-Zamelczyk, K.; Livshits, L. A Novel WT1 Mutation Identified in a 46,XX Testicular/Ovotesticular DSD Patient Results in the Retention of Intron 9. Biology 2021, 10, 1248. https://doi.org/10.3390/biology10121248
Sirokha D, Gorodna O, Vitrenko Y, Zelinska N, Ploski R, Nef S, Jaruzelska J, Kusz-Zamelczyk K, Livshits L. A Novel WT1 Mutation Identified in a 46,XX Testicular/Ovotesticular DSD Patient Results in the Retention of Intron 9. Biology. 2021; 10(12):1248. https://doi.org/10.3390/biology10121248
Chicago/Turabian StyleSirokha, Dmytro, Olexandra Gorodna, Yakov Vitrenko, Nataliya Zelinska, Rafal Ploski, Serge Nef, Jadwiga Jaruzelska, Kamila Kusz-Zamelczyk, and Ludmila Livshits. 2021. "A Novel WT1 Mutation Identified in a 46,XX Testicular/Ovotesticular DSD Patient Results in the Retention of Intron 9" Biology 10, no. 12: 1248. https://doi.org/10.3390/biology10121248
APA StyleSirokha, D., Gorodna, O., Vitrenko, Y., Zelinska, N., Ploski, R., Nef, S., Jaruzelska, J., Kusz-Zamelczyk, K., & Livshits, L. (2021). A Novel WT1 Mutation Identified in a 46,XX Testicular/Ovotesticular DSD Patient Results in the Retention of Intron 9. Biology, 10(12), 1248. https://doi.org/10.3390/biology10121248