
Metabolic Reprogramming, Questioning, and Implications for Cancer


{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
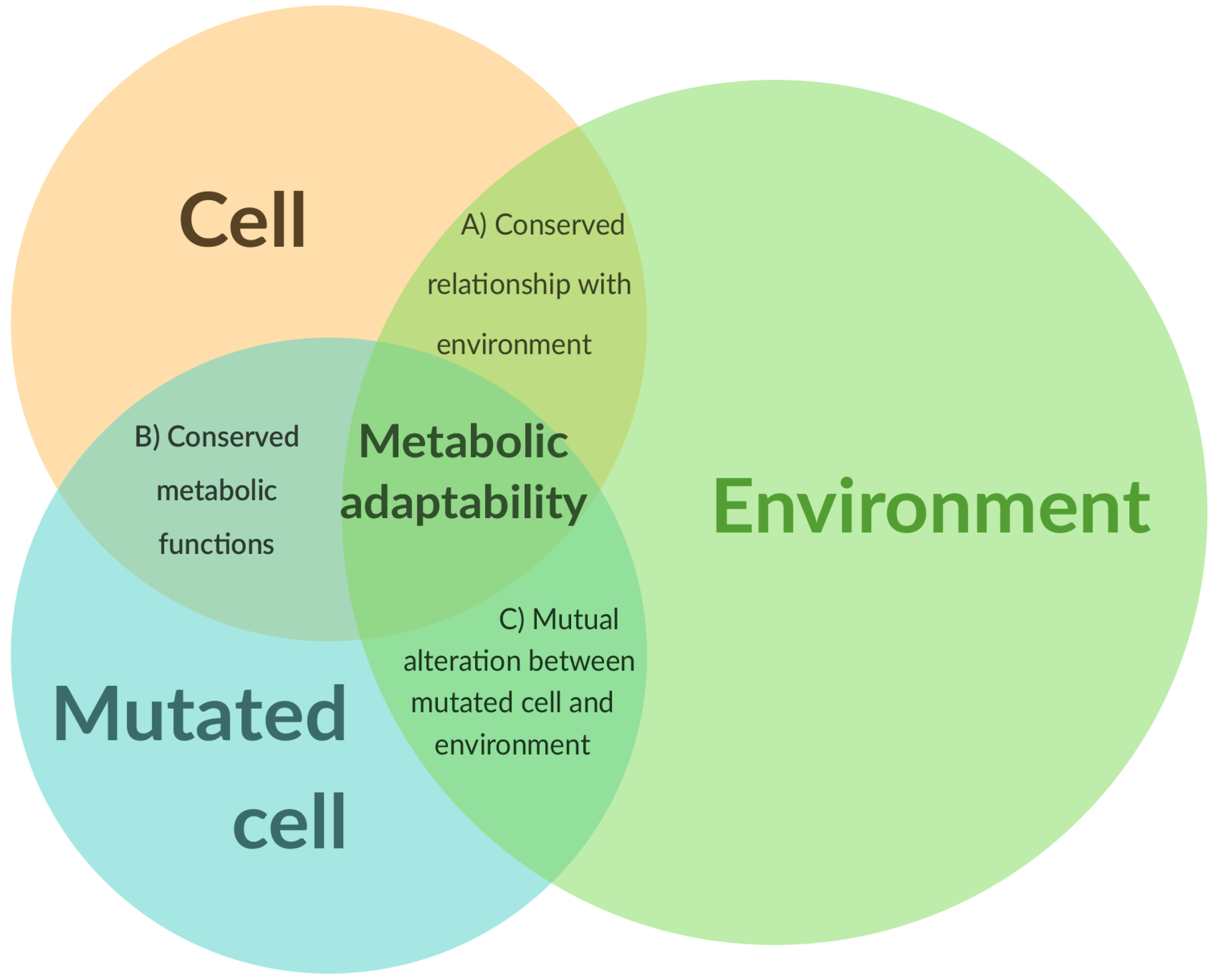
2. Is There One Metabolic Signature That Distinguishes a Normal and Tumor Phenotype?
3. How Important Is the Influence of the Tumor Heterogeneity on the Metabolism at the Tissue Scale?
4. Reprogramming or Adaptability?
- The different metabolic scenarios are already part of the “program” as conditional blocks (if–else blocks); therefore, there is no “reprogramming”.
- Parts of the code are randomly added or deleted, such as random mutations more akin to gain/loss of functions. This is following a classic evolutionary mechanism and not an in-depth transformation of the program itself.
- The program and its structure are changed deterministically in an optimal way and in a single attempt (copy–paste). Such determinism that evades trials and errors processes is not documented in biology.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhou, J.; Sun, H.; Qiu, Z.; Gao, X.; Xu, Y. CaMeRe: A Novel Tool for Inference of Cancer Metabolic Reprogramming. Front. Oncol. 2020, 10, 207. [Google Scholar] [CrossRef]
- Cazzaniga, M.; Bonanni, B. Relationship between metabolic reprogramming and mitochondrial activity in cancer cells. Understanding the anticancer effect of metformin and its clinical implications. Anticancer Res. 2015, 35, 5789–5796. [Google Scholar] [PubMed]
- Carvalho-Santos, Z.; Cardoso-Figueiredo, R.; Elias, A.P.; Tastekin, I.; Baltazar, C.; Ribeiro, C. Cellular metabolic reprogramming controls sugar appetite in Drosophila. Nat. Metab. 2020, 2, 958–973. [Google Scholar] [CrossRef]
- Konopka, A.K. Grand metaphors of biology in the genome era. Comput. Chem. 2002, 26, 397–401. [Google Scholar] [CrossRef]
- McLeod, C.; Nerlich, B. Synthetic biology, metaphors and responsibility. Life Sci. Soc. Policy 2017, 13, 13. [Google Scholar] [CrossRef] [Green Version]
- Boudry, M.; Pigliucci, M. The mismeasure of machine: Synthetic biology and the trouble with engineering metaphors. Stud. Hist. Philos. Sci. Part C Stud. Hist. Philos. Biol. Biomed. Sci. 2013, 44, 660–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Wu, H.; Dai, C.; Pan, Q.; Ding, Z.; Hu, D.; Ji, B.; Luo, Y.; Hu, X. Beyond Warburg effect - Dual metabolic nature of cancer cells. Sci. Rep. 2014, 4, 4927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, C. Metabolism and cancer: The future is now. Br. J. Cancer 2020, 122, 133–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.T.; Huang, C.H.; Wei, Y.Q. The metabolic switch and its regulation in cancer cells. Sci. China Life Sci. 2010, 53, 942–958. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; O’Neill, L.A. How should we talk about metabolism? Nat. Immunol. 2020, 21, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Dienel, G.A.; Cruz, N.F. Aerobic glycolysis during brain activation: Adrenergic regulation and influence of norepinephrine on astrocytic metabolism. J. Neurochem. 2016, 138, 14–52. [Google Scholar] [CrossRef] [Green Version]
- Jones, W.; Bianchi, K. Aerobic glycolysis: Beyond proliferation. Front. Immunol. 2015, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, M.P.; Bintener, T.; Ternes, D.; Kulms, D.; Haan, S.; Letellier, E.; Sauter, T. Identifying and targeting cancer-specific metabolism with network-based drug target prediction. EBioMedicine 2019, 43, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. pH sensing and regulation in cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [Green Version]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Yang, H.; Wang, H.; Ye, Z.; Zhou, Z.; Gu, L.; Chen, J.; Xiao, Y.; Liang, X.; Qian, X.; et al. Highly Sensitive Hill-Type Small-Molecule pH Probe That Recognizes the Reversed pH Gradient of Cancer Cells. Anal. Chem. 2018, 90, 5803–5809. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Targeted Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Strickaert, A.; Saiselet, M.; Dom, G.; De Deken, X.; Dumont, J.E.; Feron, O.; Sonveaux, P.; Maenhaut, C. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 2017, 36, 2637–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiani, C.; Maspero, D.; Di Filippo, M.; Colombo, R.; Pescini, D.; Graudenzi, A.; Westerhoff, H.V.; Alberghina, L.; Vanoni, M.; Mauri, G. Integration of single-cell RNA-seq data into population models to characterize cancer metabolism. PLoS Comput. Biol. 2019, 15, e1006733. [Google Scholar] [CrossRef]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Dewhirst, M.W. Relationships between cycling hypoxia, HIF-1, angiogenesis and oxidative stress. Radiat. Res. 2009, 172, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Yeung, S.C.J.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Orang, A.V.; Petersen, J.; McKinnon, R.A.; Michael, M.Z. Micromanaging aerobic respiration and glycolysis in cancer cells. Mol. Metab. 2019, 23, 98–126. [Google Scholar] [CrossRef]
- Morrish, F.; Neretti, N.; Sedivy, J.M.; Hockenbery, D.M. The oncogene c-Myc coordinates regulation of metabolic networks to enable rapid cell cycle entry. Cell Cycle 2008, 7, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Finley, L.W. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Kumareswaran, R.; Ludkovski, O.; Meng, A.; Sykes, J.; Pintilie, M.; Bristow, R.G. Chronic hypoxia compromises repair of DNA double-strand breaks to drive genetic instability. J. Cell Sci. 2012, 125, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Medina, M.Á. Metabolic Reprogramming is a Hallmark of Metabolism Itself. BioEssays 2020, 42, 2000058. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Lastraioli, E.; Iorio, J.; Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2685–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wang, J. Uncovering the Underlying Mechanisms of Cancer Metabolism through the Landscapes and Probability Flux Quantifications. iScience 2020, 23, 101002. [Google Scholar] [CrossRef] [PubMed]
- Yugi, K.; Kuroda, S. Metabolism-Centric Trans-Omics. Cell Syst. 2017, 4, 19–20. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquet, P.; Stéphanou, A. Metabolic Reprogramming, Questioning, and Implications for Cancer. Biology 2021, 10, 129. https://doi.org/10.3390/biology10020129
Jacquet P, Stéphanou A. Metabolic Reprogramming, Questioning, and Implications for Cancer. Biology. 2021; 10(2):129. https://doi.org/10.3390/biology10020129
Chicago/Turabian StyleJacquet, Pierre, and Angélique Stéphanou. 2021. "Metabolic Reprogramming, Questioning, and Implications for Cancer" Biology 10, no. 2: 129. https://doi.org/10.3390/biology10020129
APA StyleJacquet, P., & Stéphanou, A. (2021). Metabolic Reprogramming, Questioning, and Implications for Cancer. Biology, 10(2), 129. https://doi.org/10.3390/biology10020129