
RhoA- and Actin-Dependent Functions of Macrophages from the Rodent Cardiac Transplantation Model Perspective -Timing Is the Essence



 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
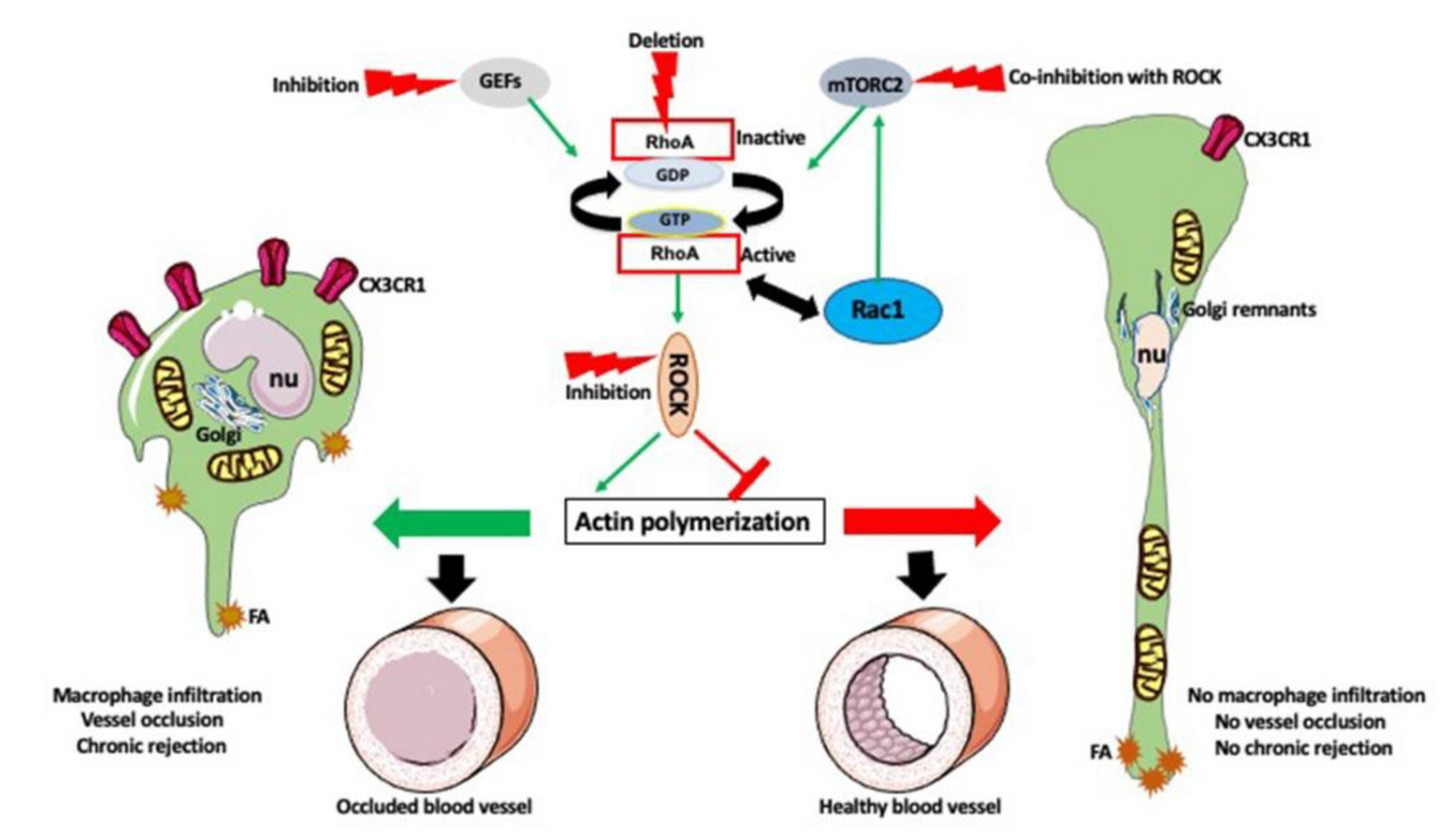
2. Actin-Dependent Functions of the Macrophages
3. Phagocytosis
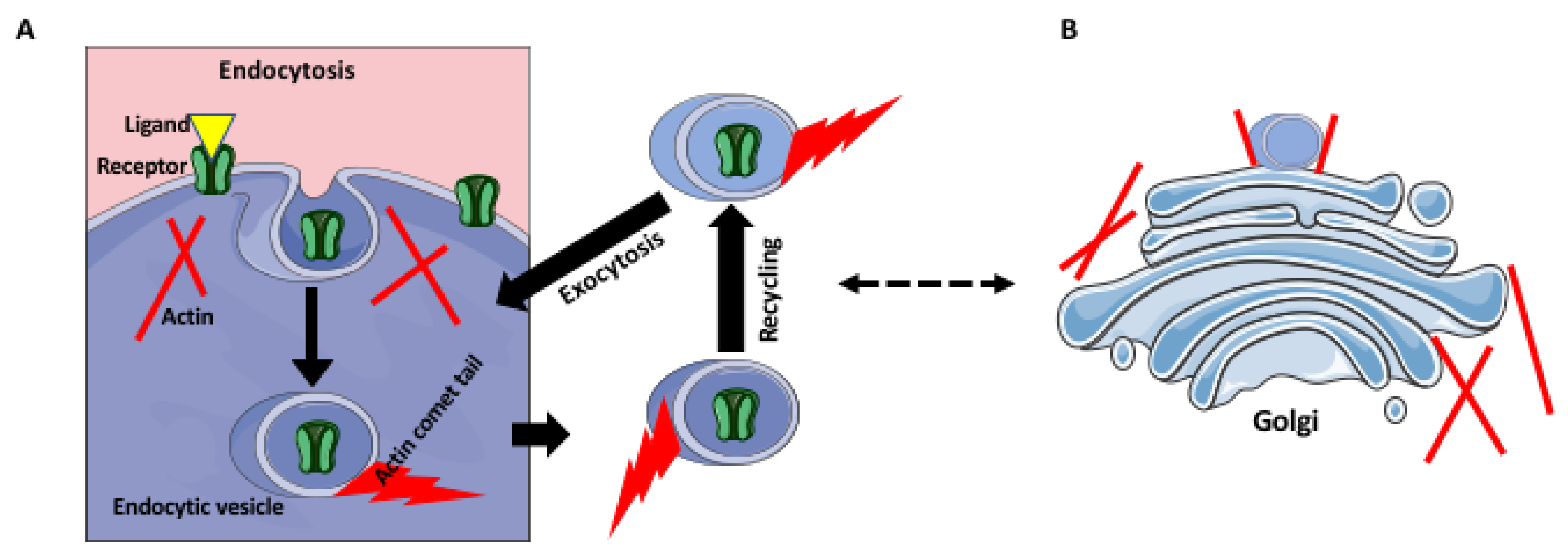
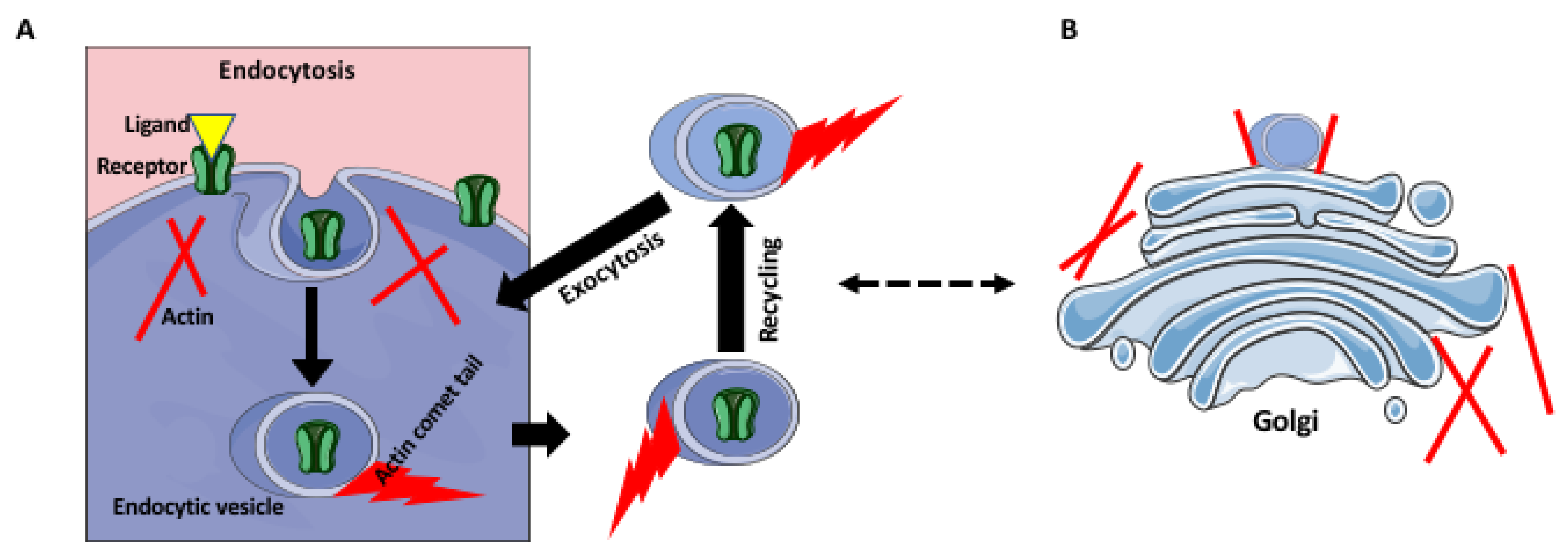
4. Receptor Trafficking and Recycling
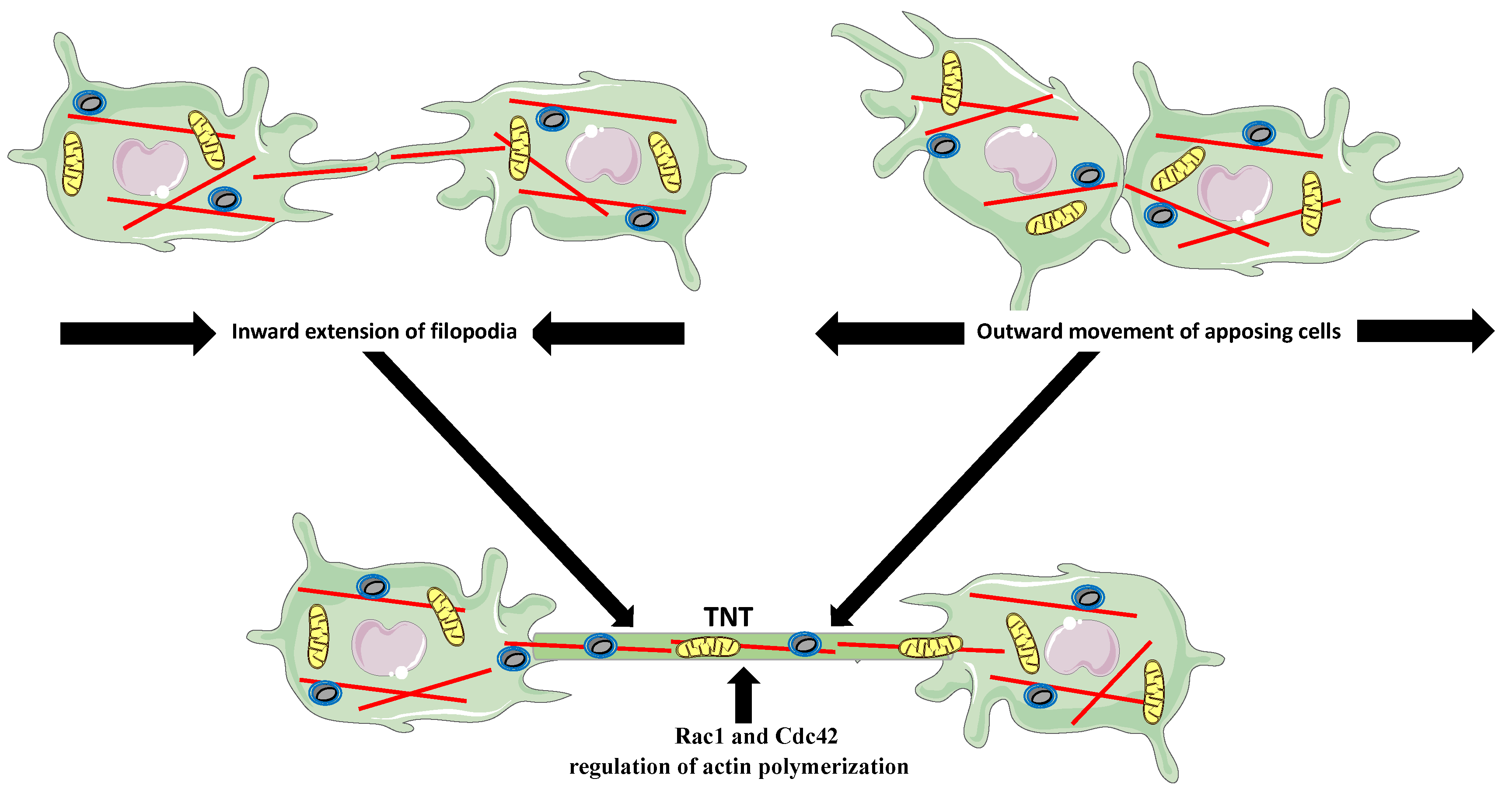
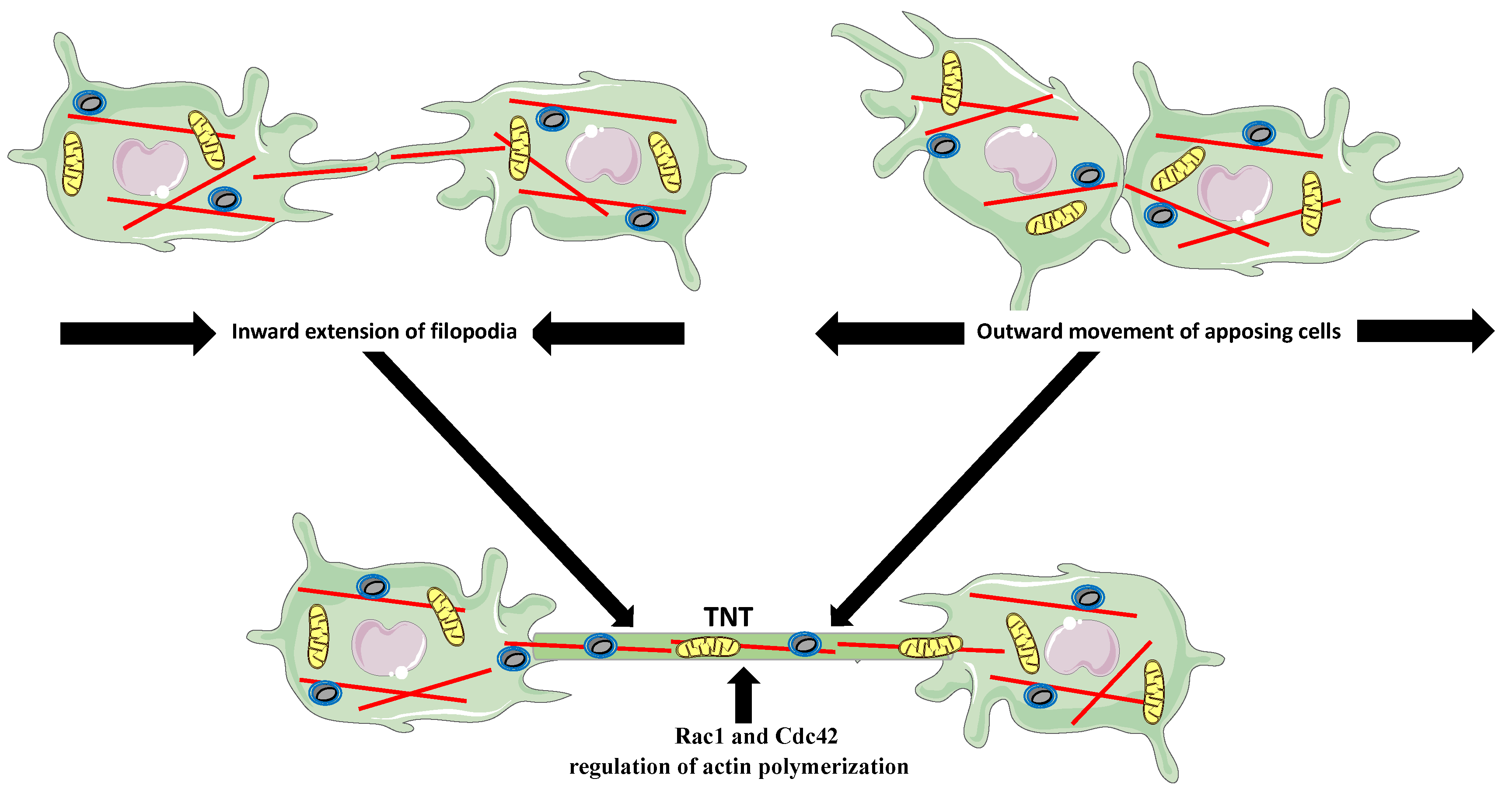
5. Tunneling Nanotubes (TNTs)
6. Cell Morphology and Locomotion
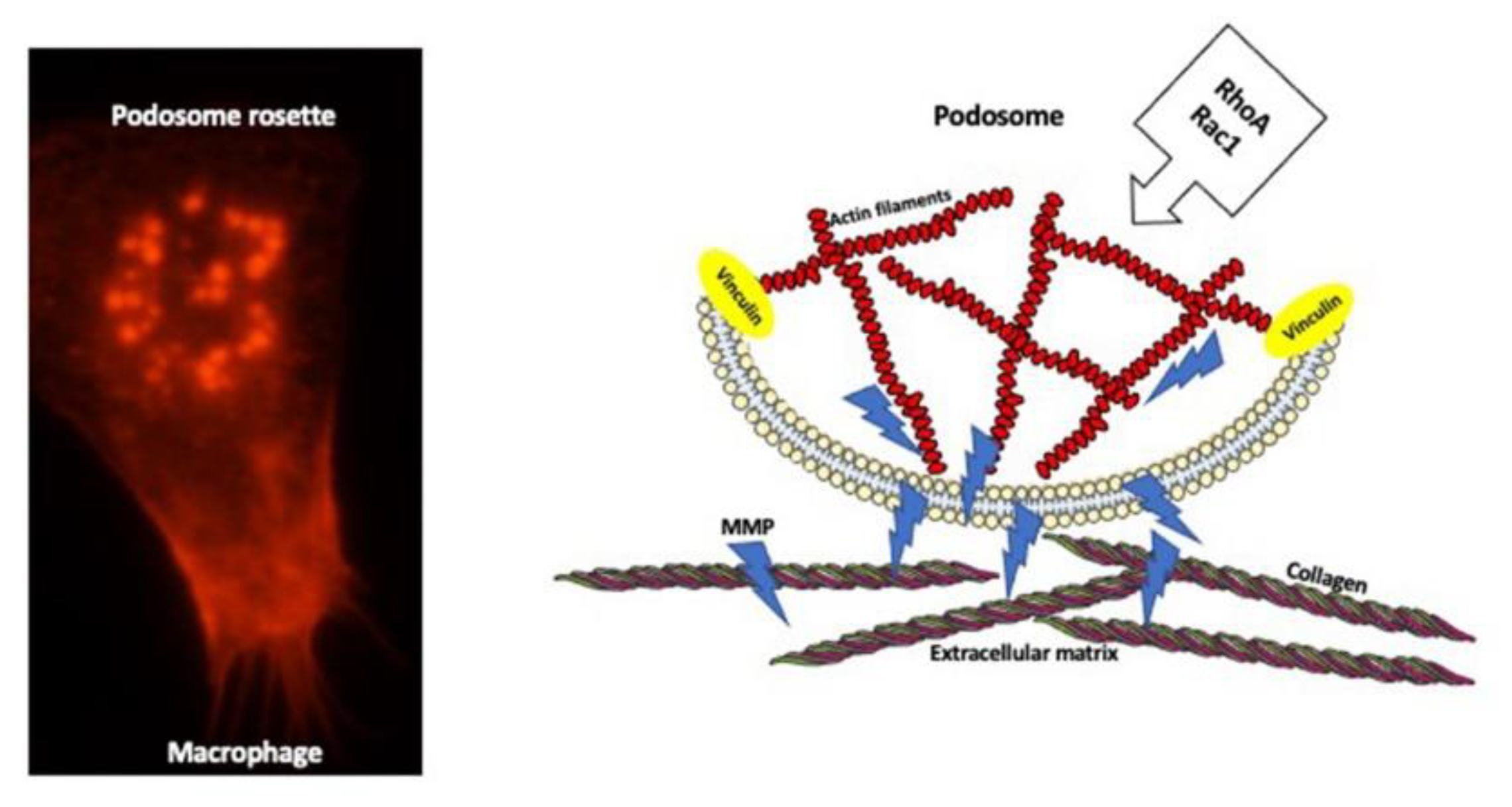
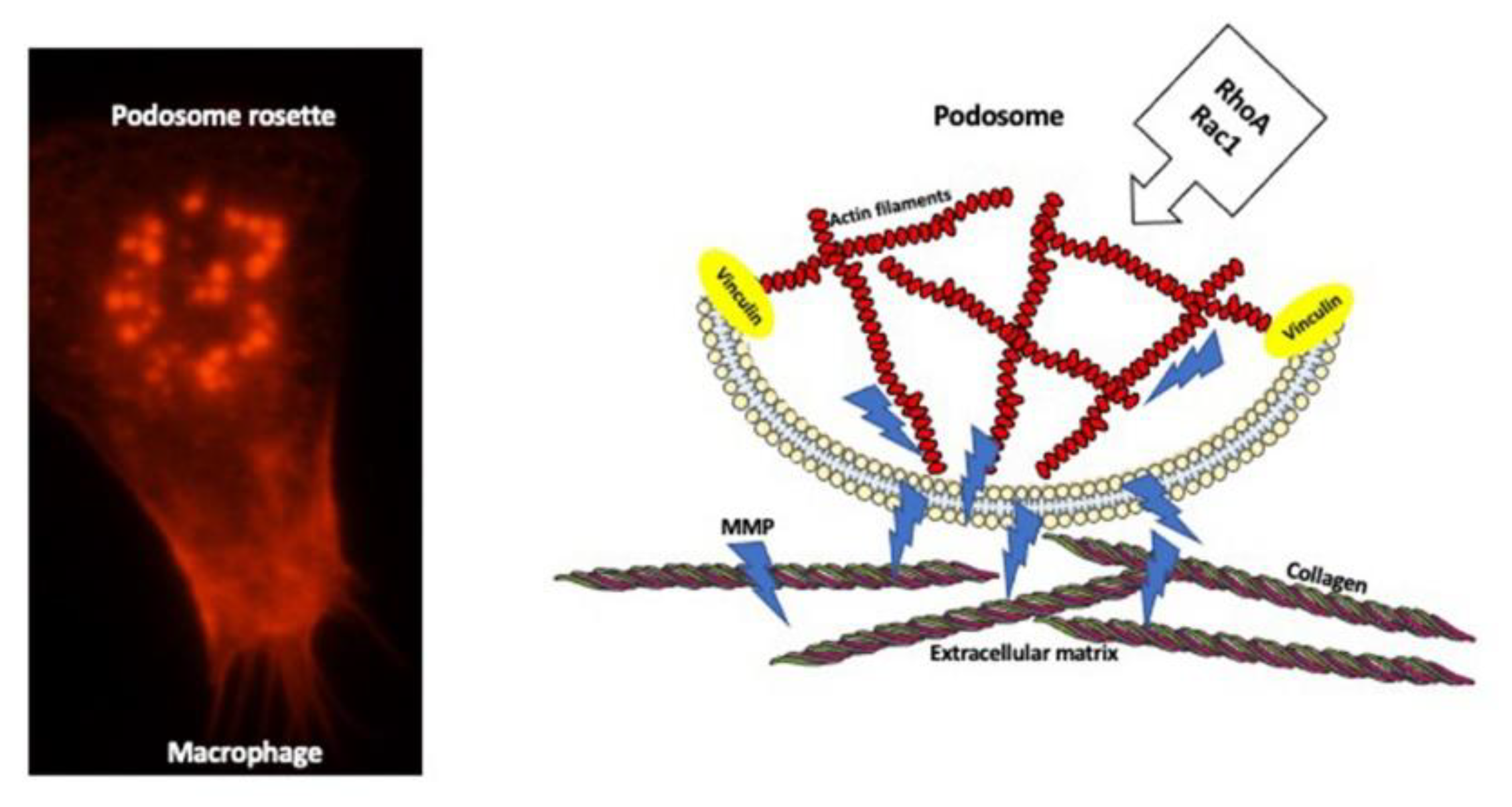
7. Extracellular Matrix Degradation
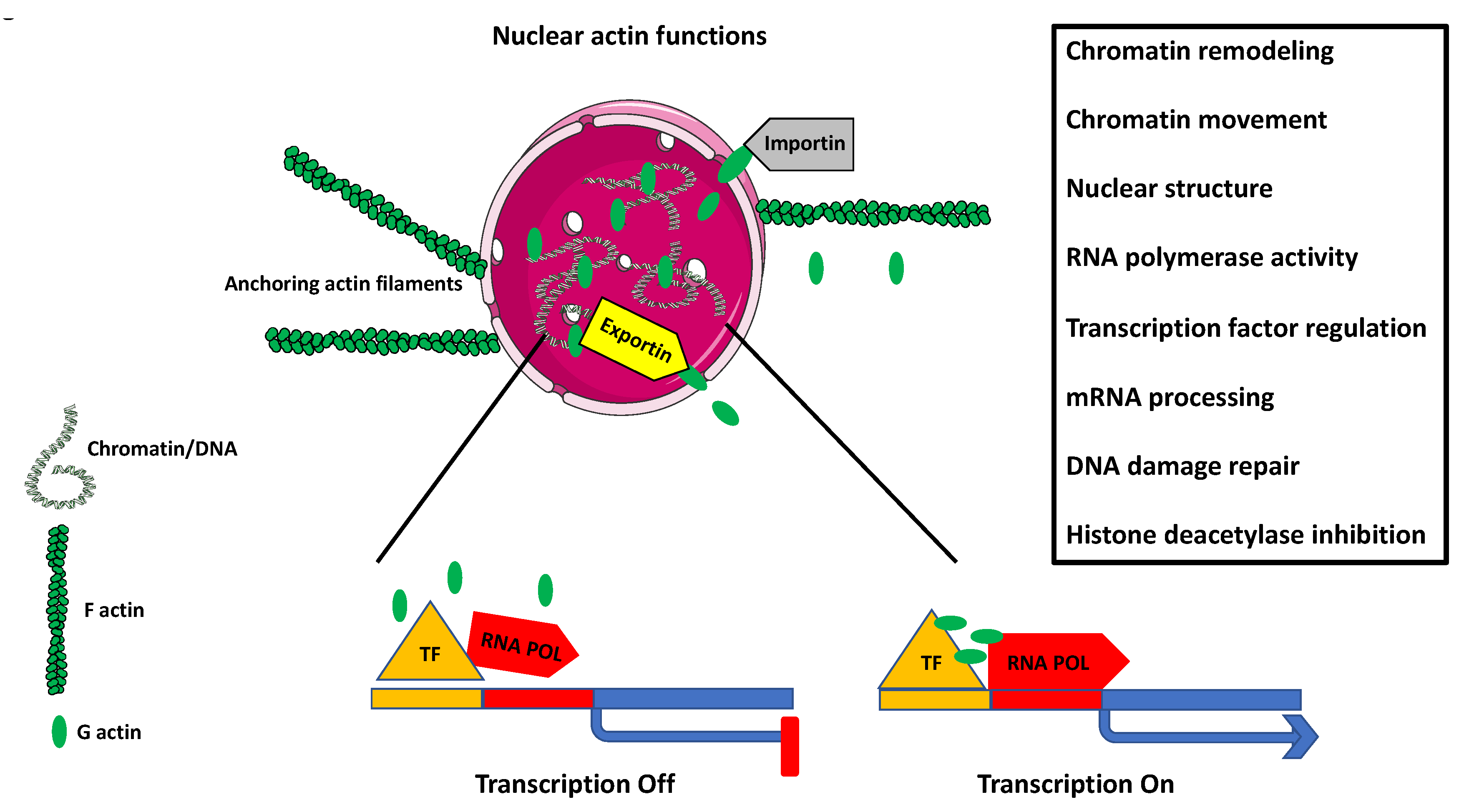
8. Nuclear Actin
9. Transplantation Model and Methods to Study Macrophages
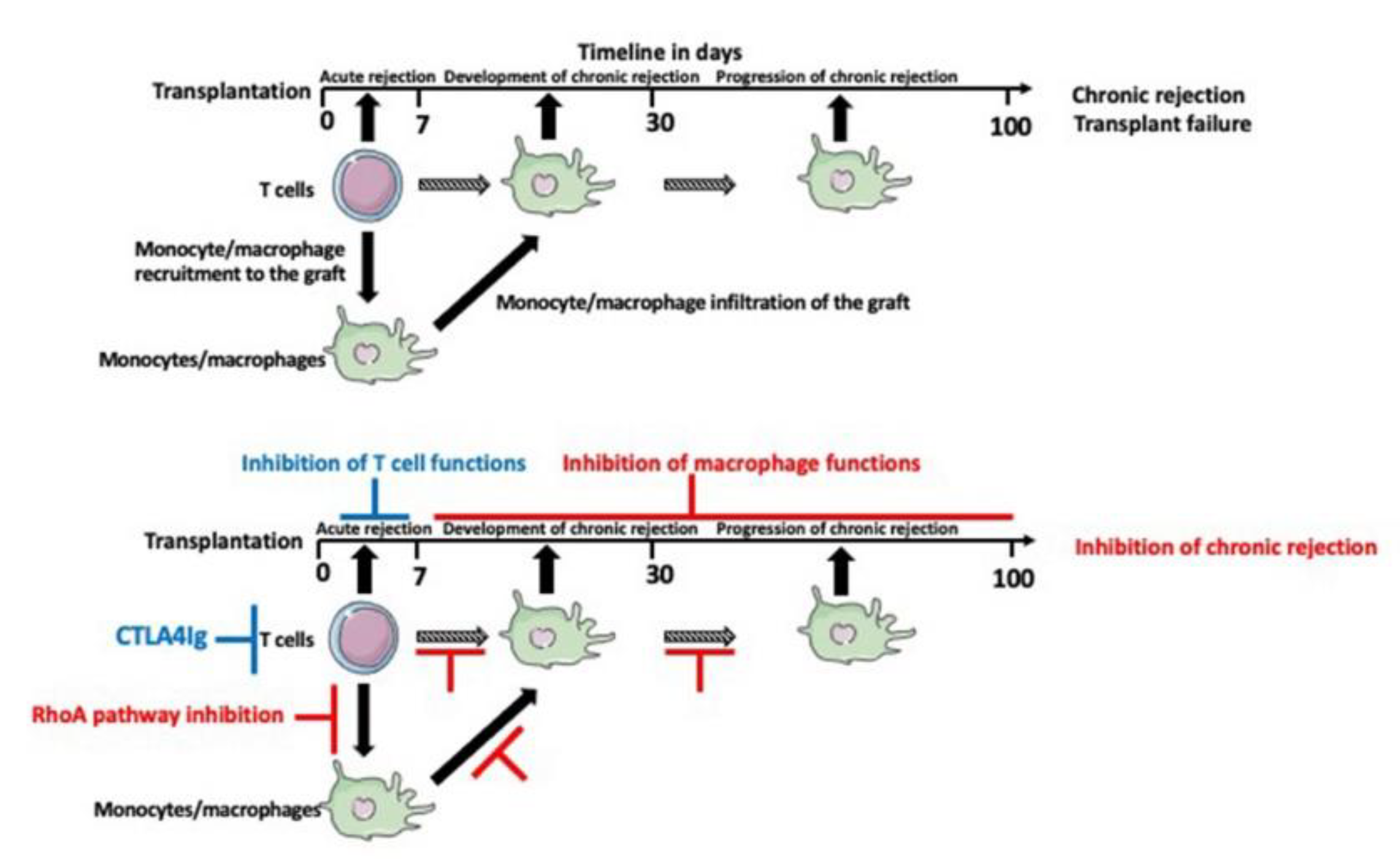
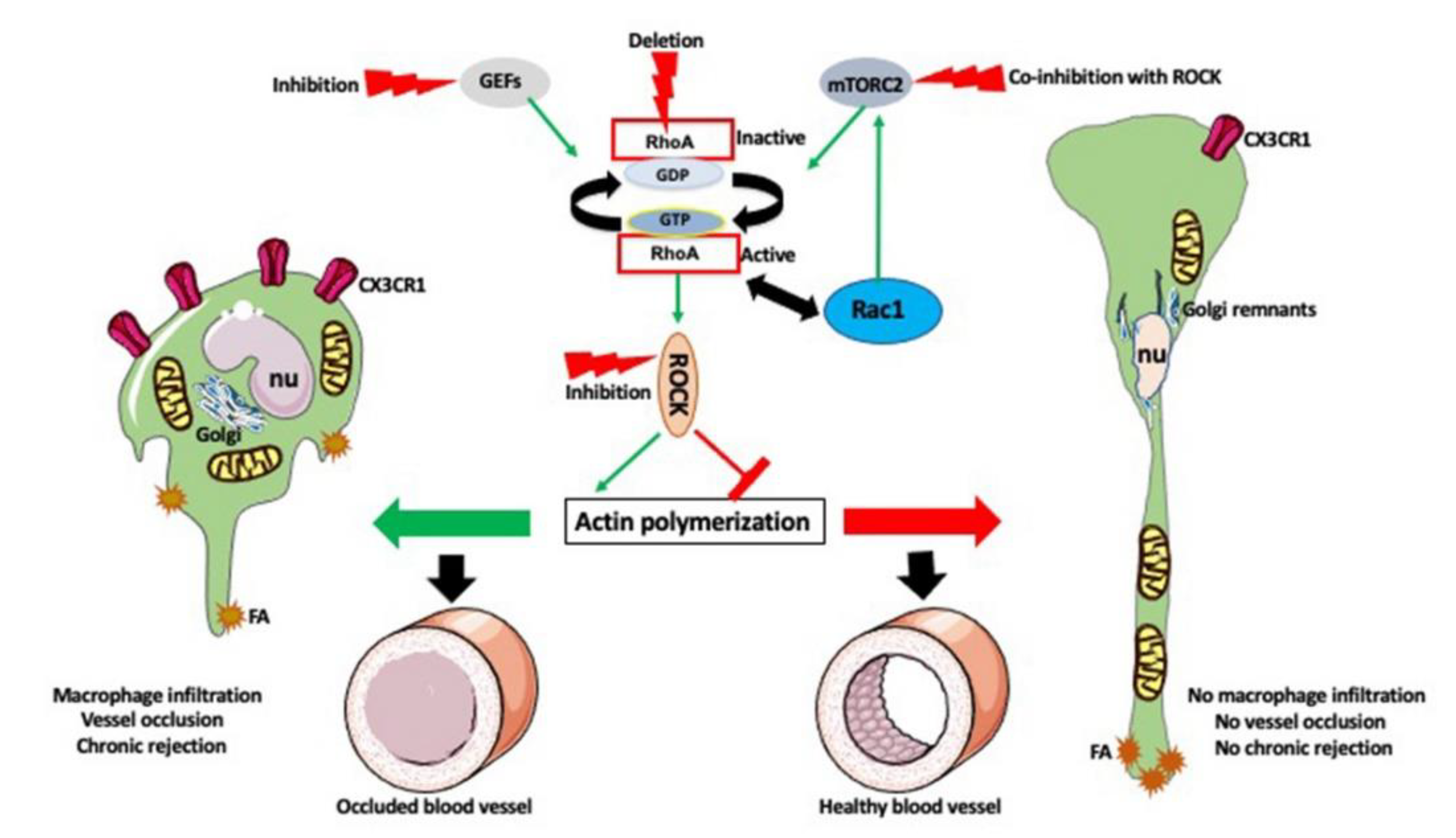
10. Timing Is the Essence
11. Conclusions and Future Approaches
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Justiz Vaillant, A.A.; Mohseni, M. Chronic Transplantation Rejection. In StatPearls; StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar] [PubMed]
- Gunaratnam, L.; Jevnikar, A.M.; Mannon, R.B. Small Animal Models of Transplantation. In Textbook of Organ Transplantation; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 158–184. [Google Scholar]
- Liu, Y.; Kloc, M.; Li, X.C. Macrophages as Effectors of Acute and Chronic Allograft Injury. Curr. Transplant. Rep. 2016, 3, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, W.; Wu, C.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Kloc, M.; Ghobrial, R.M. Macrophage/monocyte-specific deletion of Ras homolog gene family member A (RhoA) downregulates fractalkine receptor and inhibits chronic rejection of mouse cardiac allografts. J. Hear. Lung Transplant. 2017, 36, 340–354. [Google Scholar] [CrossRef] [Green Version]
- Kierdorf, K.; Prinz, M.; Geissmann, F.; Perdiguero, E.G. Development and function of tissue resident macrophages in mice. Semin. Immunol. 2015, 27, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Perdiguero, E.G.; Geissmann, F. The development and maintenance of resident macrophages. Nat. Immunol. 2016, 17, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloc, M.; Li, X.C.; Ghobrial, R.M. RhoA cytoskeletal pathway to transplantation. J. Immunol. Clin. Res. 2014, 2, 1012–2014. [Google Scholar]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, R.C.; Machesky, L.M. Phagocytosis and the actin cytoskeleton. J. Cell Sci. 2001, 114, 1061–1077. [Google Scholar] [PubMed]
- Chimini, G.; Chavrier, P. Function of Rho family proteins in actin dynamics during phagocytosis and engulfment. Nat. Cell Biol. 2000, 2, E191–E196. [Google Scholar] [CrossRef]
- Aguilera, M.; Salinas, R.; Rosales, E.; Carminati, S.; Colombo, M.I.; Berón, W. Actin Dynamics and Rho GTPases Regulate the Size and Formation of Parasitophorous Vacuoles Containing Coxiella burnetii. Infect. Immun. 2009, 77, 4609–4620. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, F.; Brumell, J.; Al-Alawi, N.; Latour, S.; Cheng, A.; Veillette, A.; Grinstein, S.; Pawson, T. The Syk protein tyrosine kinase is essential for Fc gamma receptor signaling in macrophages and neutrophils. Mol. Cell. Biol. 1998, 18, 4209–4220. [Google Scholar] [CrossRef] [Green Version]
- Crowley, M.T.; Costello, P.S.; Fitzer-Attas, C.J.; Turner, M.; Meng, F.; Lowell, C.; Tybulewicz, V.L.; DeFranco, A.L. A critical role for Syk in signal transduction and phagocytosis mediated by Fc gamma receptors on macrophages. J. Exp. Med. 1997, 186, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by Rho GTPases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Southwick, F.S.; Purich, D.L. Actin-based phagosome motility. Cell Motil. Cytoskelet. 2002, 53, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Merrifield, C.J.; Moss, S.E.; Ballestrem, C.; Imhof, B.A.; Giese, G.; Wunderlich, I.; Almers, W. Endocytic vesicles move at the tips of actin tails in cultured mast cells. Nat. Cell Biol. 1999, 1, 72–74. [Google Scholar] [CrossRef]
- Rozelle, A.L.; Machesky, L.M.; Yamamoto, M.; Driessens, M.H.; Insall, R.H.; Roth, M.G.; Luby-Phelps, K.; Marriott, G.; Hall, A.; Yin, H.L. Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr. Biol. 2000, 10, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Tanaka, M.; Okabe, Y.; Hanayama, R.; Nagata, S. Opposite Effects of Rho Family GTPases on Engulfment of Apoptotic Cells by Macrophages. J. Biol. Chem. 2006, 281, 8836–8842. [Google Scholar] [CrossRef] [Green Version]
- Tosello-Trampont, A.-C.; Nakada-Tsukui, K.; Ravichandran, K.S. Engulfment of Apoptotic Cells Is Negatively Regulated by Rho-mediated Signaling. J. Biol. Chem. 2003, 278, 49911–49919. [Google Scholar] [CrossRef] [Green Version]
- Hanley, J.G. Actin-dependent mechanisms in AMPA receptor trafficking. Front. Cell. Neurosci. 2014, 8, 381. [Google Scholar] [CrossRef] [Green Version]
- Galletta, B.J.; Cooper, J.A. Actin and endocytosis: Mechanisms and phylogeny. Curr. Opin. Cell Biol. 2009, 21, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Mooren, O.L.; Galletta, B.J.; Cooper, J.A. Roles for Actin Assembly in Endocytosis. Annu. Rev. Biochem. 2012, 81, 661–686. [Google Scholar] [CrossRef]
- Porat-Shliom, N.; Milberg, O.; Masedunskas, A.; Weigert, R. Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell. Mol. Life Sci. 2013, 70, 2099–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxfield, F.R.; McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004, 5, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.D.; Donaldson, J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009, 10, 597–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Progida, C.; Bakke, O. Bidirectional traffic between the Golgi and the endosomes–machineries and regulation. J. Cell Sci. 2016, 129, 3971–3982. [Google Scholar] [CrossRef] [Green Version]
- Kloc, M.; Uosef, A.; Wosik, J.; Kubiak, J.Z.; Ghobrial, R.M. RhoA Pathway and Actin Regulation of the Golgi/Centriole Complex. Results Probl Cell Differ. 2019, 67, 81–93. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Hanna, S.J.; McCoy-Simandle, K.; Miskolci, V.; Guo, P.; Cammer, M.; Hodgson, L.; Cox, D. The Role of Rho-GTPases and actin polymerization during Macrophage Tunneling Nanotube Biogenesis. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Hanna, S.J.; McCoy-Simandle, K.; Leung, E.; Genna, A.; Condeelis, J.S.; Cox, D. Tunneling nanotubes, a novel mode of tumor cell–macrophage communication in tumor cell invasion. J. Cell Sci. 2019, 132, jcs223321. [Google Scholar] [CrossRef] [Green Version]
- Kloc, M.; Kubiak, J.Z. Exogenous Molecule and Organelle Delivery in Oogenesis. Neurotrans. Interact. Cognit. Funct. 2017, 63, 3–16. [Google Scholar] [CrossRef]
- Kloc, M.; Kubiak, J.Z.; Bilinski, S.M. Gametic synapses, nanotubes and sperm RNAs–Redefining the origin of maternal determinants. Mech. Dev. 2016, 141, 1–3. [Google Scholar] [CrossRef]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Önfelt, B.; Nedvetzki, S.; Yanagi, K.; Davis, D.M. Cutting Edge: Membrane Nanotubes Connect Immune Cells. J. Immunol. 2004, 173, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Önfelt, B.; Nedvetzki, S.; Benninger, R.K.P.; Purbhoo, M.A.; Sowinski, S.; Hume, A.N.; Seabra, M.C.; Neil, M.A.A.; French, P.M.W.; Davis, D.M. Structurally Distinct Membrane Nanotubes between Human Macrophages Support Long-Distance Vesicular Traffic or Surfing of Bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, S.C.; Salter, R.D. Functional Connectivity between Immune Cells Mediated by Tunneling Nanotubules. Immunity 2005, 23, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, F.Y.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [Green Version]
- Schakenraad, K.; Ernst, J.; Pomp, W.; Danen, E.H.J.; Merks, R.M.H.; Schmidt, T.; Giomi, L. Mechanical interplay between cell shape and actin cytoskeleton organization. Soft Matter 2020, 16, 6328–6343. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Actin, a central player in cell shape and movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef] [Green Version]
- Gundersen, G.G.; Worman, H.J. Nuclear Positioning. Cell 2013, 152, 1376–1389. [Google Scholar] [CrossRef] [Green Version]
- Bone, C.R.; Tapley, E.C.; Gorjánácz, M.; Starr, D.A. The Caenorhabditis elegans SUN protein UNC-84 interacts with lamin to transfer forces from the cytoplasm to the nucleoskeleton during nuclear migration. Mol. Biol. Cell 2014, 25, 2853–2865. [Google Scholar] [CrossRef]
- Starr, D.A.; Han, M. ANChors away: An actin based mechanism of nuclear positioning. J. Cell Sci. 2003, 116, 211–216. [Google Scholar] [CrossRef] [Green Version]
- Lei, K.; Zhang, X.; Ding, X.; Guo, X.; Chen, M.; Zhu, B.; Xu, T.; Zhuang, Y.; Xu, R.; Han, M. SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 10207–10212. [Google Scholar] [CrossRef] [Green Version]
- Kraft, L.M.; Lackner, L.L. Mitochondrial anchors: Positioning mitochondria and more. Biochem. Biophys. Res. Commun. 2018, 500, 2–8. [Google Scholar] [CrossRef]
- Frederick, R.L.; Shaw, J.M. Moving Mitochondria: Establishing Distribution of an Essential Organelle. Traffic 2007, 8, 1668–1675. [Google Scholar] [CrossRef] [Green Version]
- Dmytriyev, A.; Tkach, V.; Rudenko, O.; Bock, E.; Berezin, V.; Cytometry, A. An automatic procedure for evaluation of single cell motility. Cytom. Part A 2006, 69, 979–985. [Google Scholar] [CrossRef]
- Tkach, V.; Bock, E.; Berezin, V. The Role of RhoA in the Regulation of Cell Morphology and Motility. Cell Motil. Cytoskelet. 2005, 61, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Koenigs, V.; Jennings, R.; Vogl, T.J.; Horsthemke, M.; Bachg, A.C.; Xu, Y.; Grobe, K.; Brakebusch, C.; Schwab, A.; Baehler, M.; et al. Mouse Macrophages Completely Lacking Rho Subfamily GTPases (RhoA, RhoB, and RhoC) Have Severe Lamellipodial Retraction Defects, but Robust Chemotactic Navigation and Altered Motility. J. Biol. Chem. 2014, 289, 30772–30784. [Google Scholar] [CrossRef] [Green Version]
- Valentin, J.E.; Stewart-Akers, A.M.; Gilbert, T.W.; Badylak, S.F. Macrophage Participation in the Degradation and Remodeling of Extracellular Matrix Scaffolds. Tissue Eng. Part A 2009, 15, 1687–1694. [Google Scholar] [CrossRef] [Green Version]
- Wiesner, C.; Le-Cabec, V.; El Azzouzi, K.; Maridonneau-Parini, I.; Linder, S. Podosomes in space: Macrophage migration and matrix degradation in 2D and 3D settings. Cell Adhes. Migr. 2014, 8, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ghobrial, R.M.; Li, X.C.; Kloc, M. Inhibition of RhoA and mTORC2/Rictor by Fingolimod (FTY720) induces p21-activated kinase 1, PAK-1 and amplifies podosomes in mouse peritoneal macrophages. Immunobiology 2018, 223, 634–647. [Google Scholar] [CrossRef]
- Liu, Y.; Tejpal, N.; You, J.; Li, X.C.; Ghobrial, R.M.; Kloc, M. ROCK inhibition impedes macrophage polarity and functions. Cell. Immunol. 2016, 300, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Spuul, P.; Ciufici, P.; Veillat, V.; Leclercq, A.; Daubon, T.; Kramer, I.; Génot, E. Importance of RhoGTPases in formation, characteristics, and functions of invadosomes. Small GTPases 2014, 5, e28195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dries, K.V.D.; Nahidiazar, L.; Slotman, J.A.; Meddens, M.B.M.; Pandzic, E.; Joosten, B.; Ansems, M.; Schouwstra, J.; Meijer, A.; Steen, R.; et al. Modular actin nano-architecture enables podosome protrusion and mechanosensing. Nat. Commun. 2019, 10, 5171. [Google Scholar] [CrossRef] [Green Version]
- Weaver, A.M.; Young, M.E.; Lee, W.-L.; Cooper, J.A. Integration of signals to the Arp2/3 complex. Curr. Opin. Cell Biol. 2003, 15, 23–30. [Google Scholar] [CrossRef]
- Van Helden, S.F.G.; Oud, M.M.; Joosten, B.; Peterse, N.; Figdor, C.G.; Van Leeuwen, F.N. PGE2-mediated podosome loss in dendritic cells is dependent on actomyosin contraction downstream of the RhoA-Rho-kinase axis. J. Cell Sci. 2008, 121, 1096–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wineland, D.M.; Kelpsch, D.J.; Tootle, T.L. Multiple Pools of Nuclear Actin. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2018, 301, 2014–2036. [Google Scholar] [CrossRef] [Green Version]
- Kelpsch, D.J.; Tootle, T.L. Nuclear Actin: From Discovery to Function. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2018, 301, 1999–2013. [Google Scholar] [CrossRef] [Green Version]
- Plessner, M.; Melak, M.; Chinchilla, P.; Baarlink, C.; Grosse, R. Nuclear F-actin Formation and Reorganization upon Cell Spreading. J. Biol. Chem. 2015, 290, 11209–11216. [Google Scholar] [CrossRef] [Green Version]
- Misu, S.; Takebayashi, M.; Miyamoto, K. Nuclear Actin in Development and Transcriptional Reprogramming. Front. Genet. 2017, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Wang, W.; Rando, O.J.; Xue, Y.; Swiderek, K.; Kuo, A.; Crabtree, G.R. Rapid and Phosphoinositol-Dependent Binding of the SWI/SNF-like BAF Complex to Chromatin after T Lymphocyte Receptor Signaling. Cell 1998, 95, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Visa, N.; Percipalle, P. Nuclear Functions of Actin. Cold Spring Harb. Perspect. Biol. 2010, 2, a000620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, P.; Shen, X. Mechanisms of nuclear actin in chromatin-remodeling complexes. Trends Cell Biol. 2014, 24, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.Z.; Thuraisingam, T.; Morais, D.A.D.L.; Rola-Pleszczynski, M.; Radzioch, D. Nuclear Translocation of β-Actin Is Involved in Transcriptional Regulation during Macrophage Differentiation of HL-60 Cells. Mol. Biol. Cell 2010, 21, 811–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedges, J.F.; Kimmel, E.; Snyder, D.T.; Jerome, M.; Jutila, M.A. Solute Carrier 11A1 Is Expressed by Innate Lymphocytes and Augments Their Activation. J. Immunol. 2013, 190, 4263–4273. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Gedda, M.R.; Tiwari, N.; Singh, S.P.; Bajpai, S.; Singh, R.K. Solute carrier protein family 11 member 1 (Slc11a1) activation efficiently inhibits Leishmania donovani survival in host macrophages. J. Parasit. Dis. 2016, 41, 671–677. [Google Scholar] [CrossRef]
- Plenter, R.J.; Grazia, T.J. Murine Heterotopic Heart Transplant Technique. J. Vis. Exp. 2014, 89, e51511. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of Gene Knockout Mice. Curr. Protoc. Cell Biol. 2009, 44, 19.12.1–19.12.17. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP system: General principles to determine tissue-specific roles of target genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Progatzky, F.; Dallman, M.J.; Celso, C.L. From seeing to believing: Labelling strategies for in vivo cell-tracking experiments. Interface Focus 2013, 3, 20130001. [Google Scholar] [CrossRef] [Green Version]
- Lisik, W.; Gong, Y.; Tejpal, N.; Skelton, T.S.; Bremer, E.G.; Kloc, M.; Ghobrial, R.M. Intragraft gene expression profile associated with the induction of tolerance by allochimeric MHC I in the rat heart transplantation model. Genes 2009, 48, 8–19. [Google Scholar] [CrossRef]
- Chen, W.; Chen, S.; Chen, W.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Coinhibition of mTORC1/mTORC2 and RhoA/ROCK pathways prevents chronic rejection of rat cardiac allografts. Transplant. Rep. 2018, 3, 21–28. [Google Scholar] [CrossRef]
- Chen, W.; Chen, S.; Chen, W.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Screening RhoA/ROCK inhibitors for the ability to prevent chronic rejection of mouse cardiac allografts. Transpl. Immunol. 2018, 50, 15–25. [Google Scholar] [CrossRef]
- Zhang, L.; Kloc, M.; Tejpal, N.; You, J.; Cordero-Reyes, A.M.; Youker, K.A.; Ghobrial, R.M. ROCK1 inhibitor abrogates chronic rejec-tion in rat cardiac model system. Open J. Organ Transp. Surg. 2012, 2, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Hua, L.; Harmer, D.; Li, P.; Ren, G. Cre Driver Mice Targeting Macrophages. Methods Mol. Biol. 2018, 1784, 263–275. [Google Scholar] [CrossRef]
- Wosik, J.; Chen, W.; Qin, K.; Ghobrial, R.M.; Kubiak, J.Z.; Kloc, M. Magnetic Field Changes Macrophage Phenotype. Biophys. J. 2018, 114, 2001–2013. [Google Scholar] [CrossRef] [Green Version]
- Wosik, J.; Suarez-Villagran, M.; Miller, J.H.; Ghobrial, R.M.; Kloc, M. Macrophage phenotype bioengineered by magnetic, genetic, or pharmacologic interference. Immunol. Res. 2019, 67, 1–11. [Google Scholar] [CrossRef]
- Acharya, B.R.; Nestor-Bergmann, A.; Liang, X.; Gupta, S.; Duszyc, K.; Gauquelin, E.; Gomez, G.A.; Budnar, S.; Marcq, P.; Jensen, O.E.; et al. A Mechanosensitive RhoA Pathway that Protects Epithelia against Acute Tensile Stress. Dev. Cell 2018, 47, 439–452.e6. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nat. Cell Biol. 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Liu, Y.; Minze, L.J.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M.; Chen, W.; Kloc, M. Dissonant response of M0/M2 and M1 bone-marrow-derived macrophages to RhoA pathway interference. Cell Tissue Res. 2016, 366, 707–720. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, Y.; Li, X.C.; Kubiak, J.Z.; Ghobrial, R.M.; Kloc, M. Rho-specific Guanine nucleotide exchange factors (Rho-GEFs) inhibition affects macrophage phenotype and disrupts Golgi complex. Int. J. Biochem. Cell Biol. 2017, 93, 12–24. [Google Scholar] [CrossRef]
- Fehrenbacher, K.; Huckaba, T.; Yang, H.-C.; Boldogh, I.; Pon, L. Actin comet tails, endosomes and endosymbionts. J. Exp. Biol. 2003, 206, 1977–1984. [Google Scholar] [CrossRef] [Green Version]
- Dogterom, M.; Koenderink, G.H. Actin–microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.R.; Pollard, T.D. Real-Time Measurements of Actin Filament Polymerization by Total Internal Reflection Fluorescence Microscopy. Biophys. J. 2005, 88, 1387–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haspel, J.A.; Anafi, R.; Brown, M.K.; Cermakian, N.; Depner, C.; Desplats, P.; Gelman, A.E.; Haack, M.; Jelic, S.; Kim, B.S.; et al. Perfect timing: Circadian rhythms, sleep, and immunity—an NIH workshop summary. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiermann, C.; Kunisaki, Y.; Frenette, P.S. Circadian control of the immune system. Nat. Rev. Immunol. 2013, 13, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Comas, M.; Gordon, C.J.; Oliver, B.; Stow, N.W.; King, G.; Sharma, P.; Ammit, A.J.; Grunstein, R.R.; Phillips, C.L. A circadian based inflammatory response–implications for respiratory disease and treatment. Sleep Sci. Pract. 2017, 1. [Google Scholar] [CrossRef]
- Timmons, G.A.; O’Siorain, J.R.; Kennedy, O.D.; Curtis, A.M.; Early, J.O. Innate Rhythms: Clocks at the Center of Monocyte and Macrophage Function. Front. Immunol. 2020, 11, 1743. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, B.; Wang, X.; Luo, C.; Zhou, T.; Zheng, X.; Ding, J.-W. Circadian rhythm and atherosclerosis (Review). Exp. Ther. Med. 2020, 20, 1. [Google Scholar] [CrossRef]
- Cheng, B.; Anea, C.B.; Yao, L.; Chen, F.; Patel, V.; Merloiu, A.; Pati, P.; Caldwell, R.W.; Fulton, D.J.; Rudic, R.D. Tissue-intrinsic dysfunction of circadian clock confers transplant arteriosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 17147–17152. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloc, M.; Uosef, A.; Villagran, M.; Zdanowski, R.; Kubiak, J.Z.; Wosik, J.; Ghobrial, R.M. RhoA- and Actin-Dependent Functions of Macrophages from the Rodent Cardiac Transplantation Model Perspective -Timing Is the Essence. Biology 2021, 10, 70. https://doi.org/10.3390/biology10020070
Kloc M, Uosef A, Villagran M, Zdanowski R, Kubiak JZ, Wosik J, Ghobrial RM. RhoA- and Actin-Dependent Functions of Macrophages from the Rodent Cardiac Transplantation Model Perspective -Timing Is the Essence. Biology. 2021; 10(2):70. https://doi.org/10.3390/biology10020070
Chicago/Turabian StyleKloc, Malgorzata, Ahmed Uosef, Martha Villagran, Robert Zdanowski, Jacek Z. Kubiak, Jarek Wosik, and Rafik M. Ghobrial. 2021. "RhoA- and Actin-Dependent Functions of Macrophages from the Rodent Cardiac Transplantation Model Perspective -Timing Is the Essence" Biology 10, no. 2: 70. https://doi.org/10.3390/biology10020070
APA StyleKloc, M., Uosef, A., Villagran, M., Zdanowski, R., Kubiak, J. Z., Wosik, J., & Ghobrial, R. M. (2021). RhoA- and Actin-Dependent Functions of Macrophages from the Rodent Cardiac Transplantation Model Perspective -Timing Is the Essence. Biology, 10(2), 70. https://doi.org/10.3390/biology10020070