
Altered Expression of Candidate Genes in Mayer–Rokitansky–Küster–Hauser Syndrome May Influence Vaginal Keratinocytes Biology: A Focus on Protein Kinase X

 ,
,  , ,
, ,  , ,
, ,  , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Cell Cultures and Treatments
2.3. RNA Extraction, Reverse Transcription and Quantitative Real-Time PCR (RT-qPCR)
2.4. Principal Component Analysis (PCA)
2.5. PRKX Cloning and Transfection
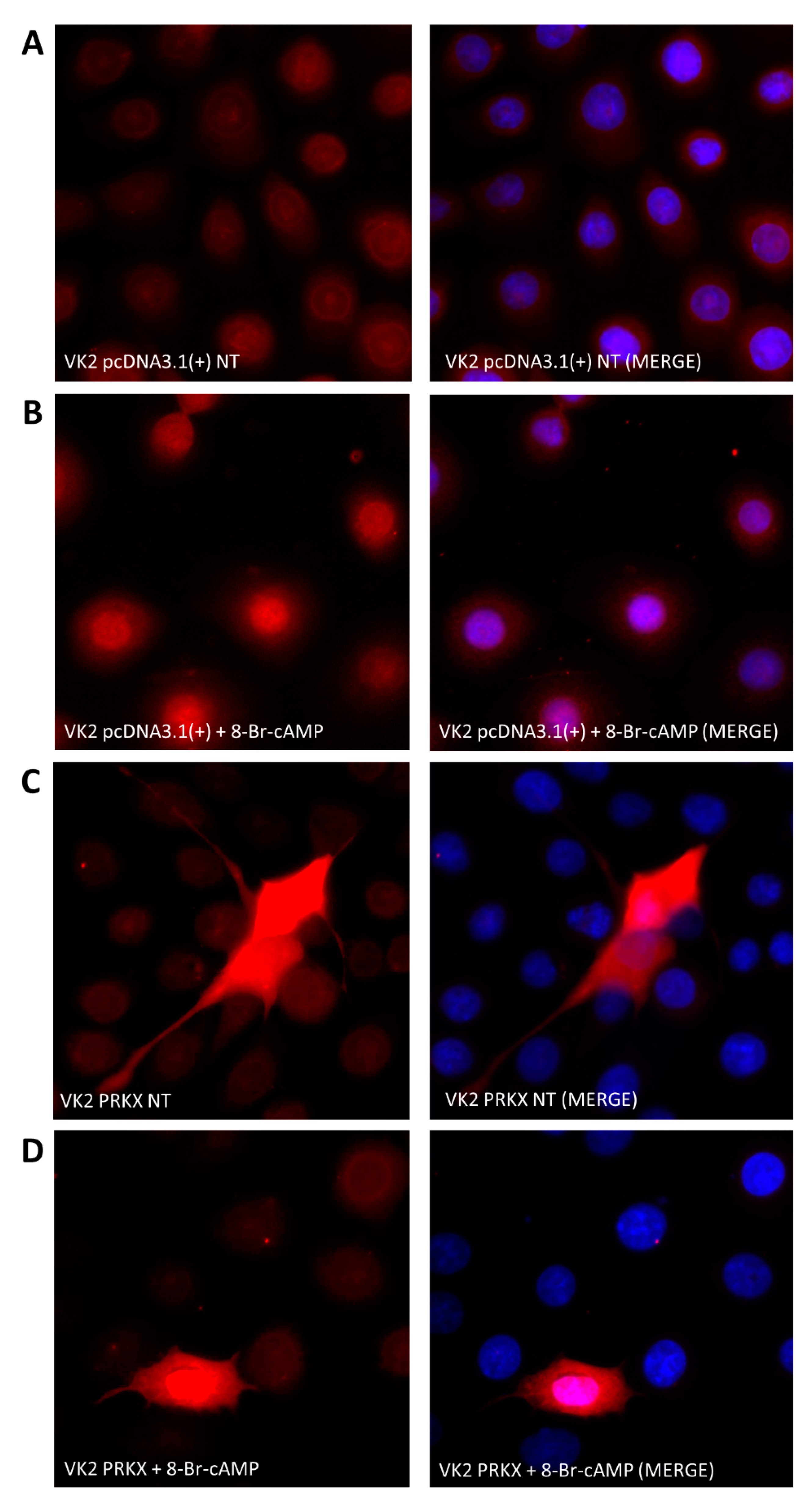
2.6. Immunofluorescence
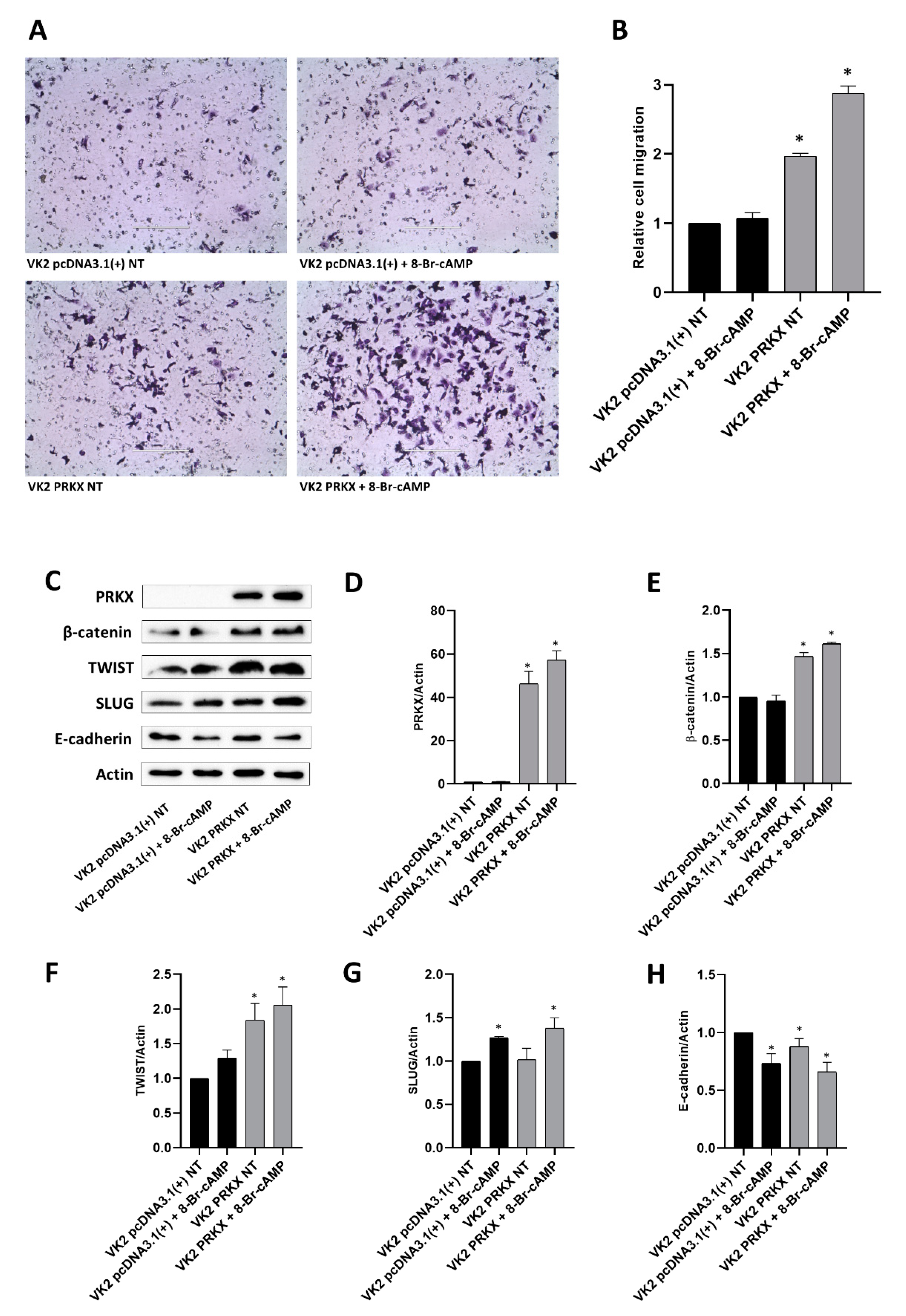
2.7. Protein Extracts and Western Blot Analysis
2.8. Cell Migration Assay
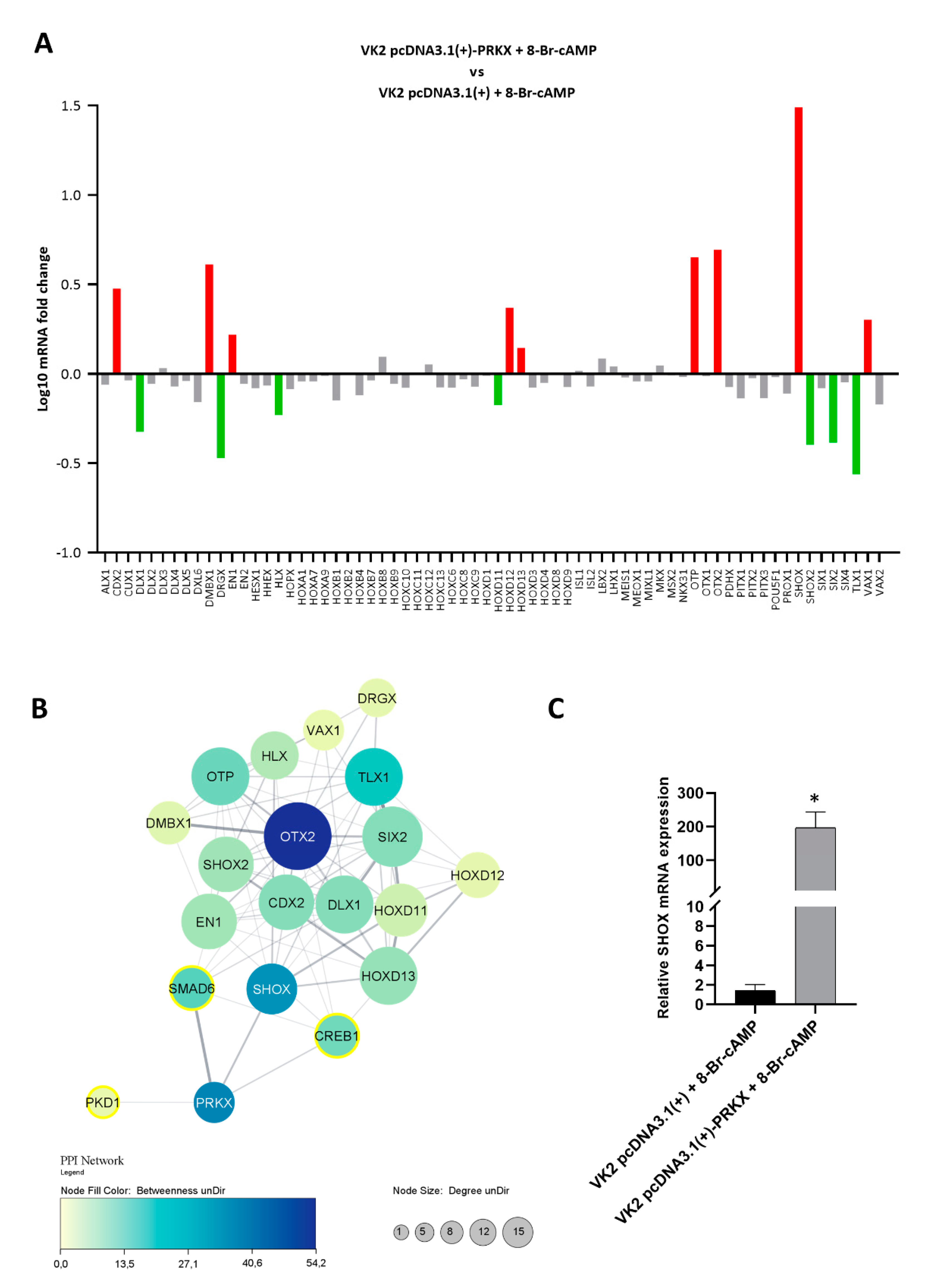
2.9. Protein–Protein Interaction (PPI) Network Analysis
2.10. Statistics
3. Results
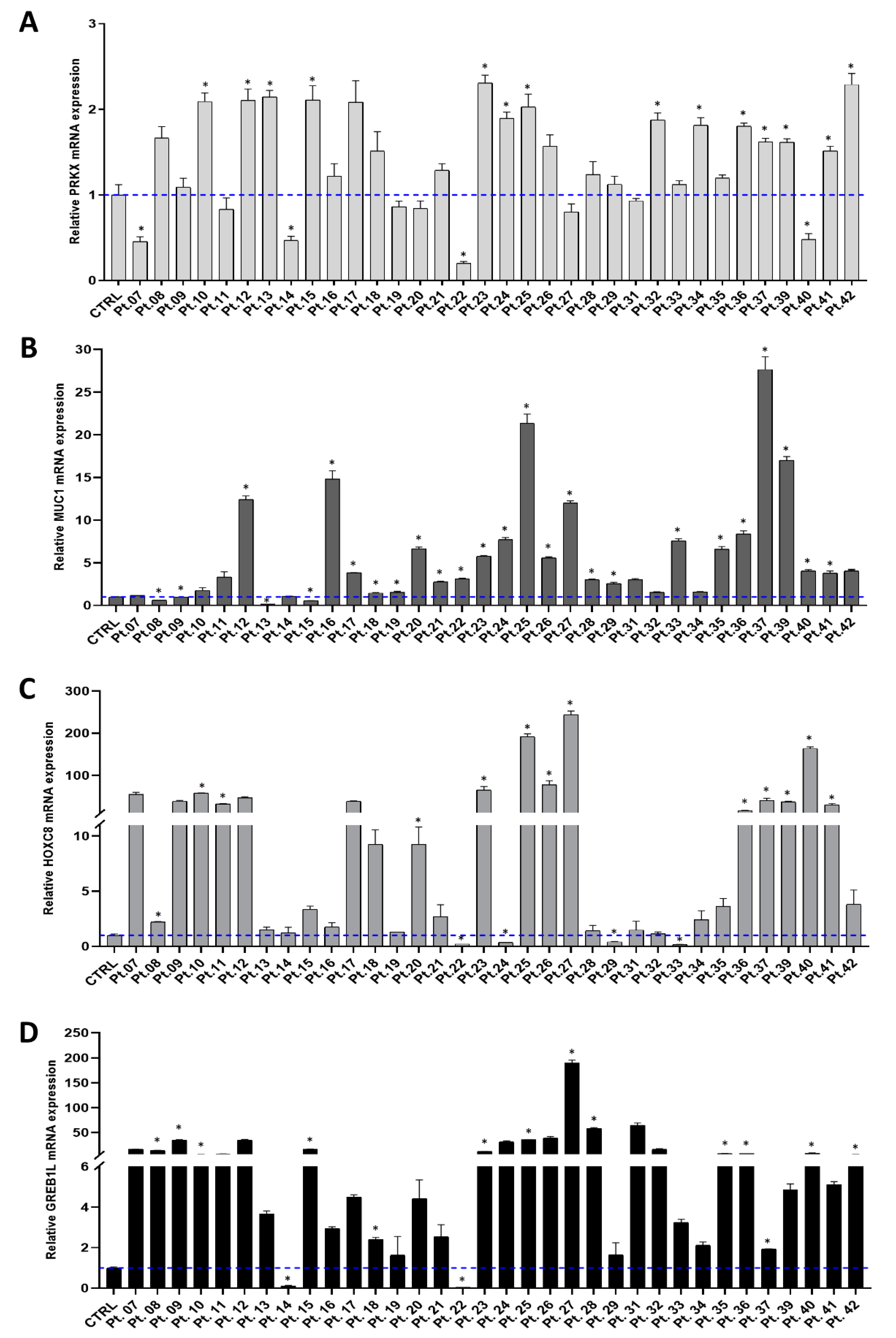
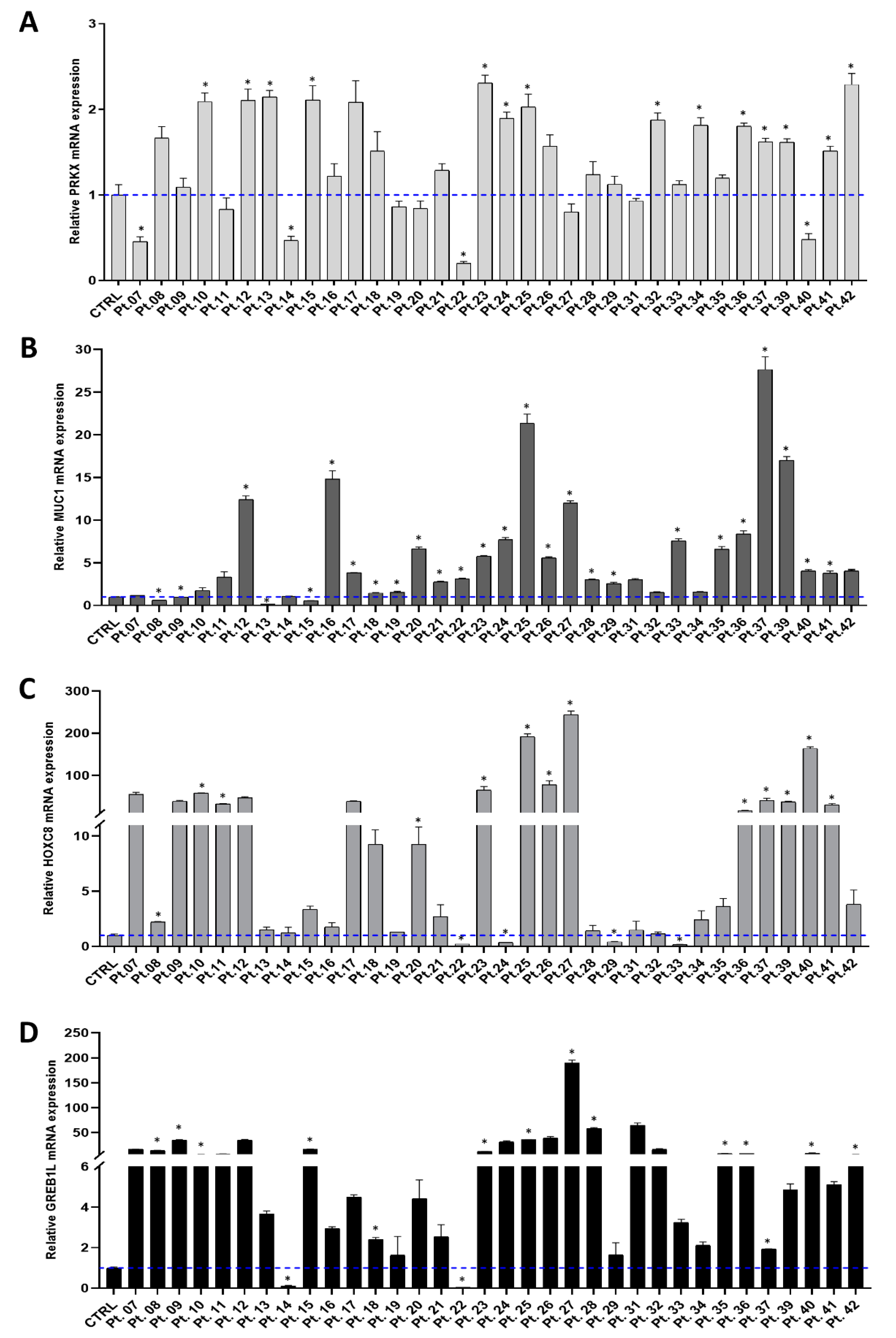
3.1. Investigating PRKX and Selected Candidate Genes Expression in MRKH Patients
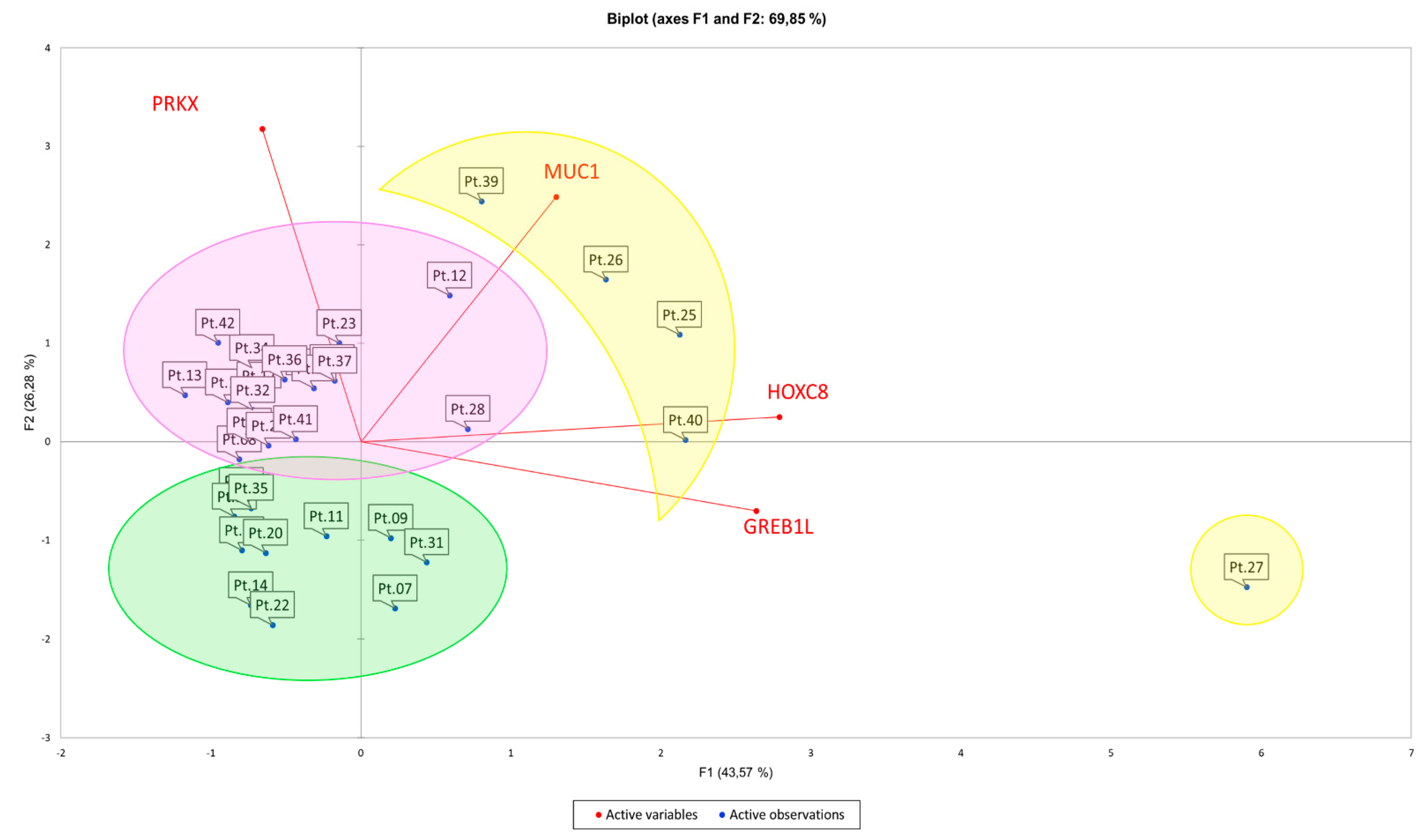
3.2. Principal Component Analysis (PCA) Suggests Correlations between Expression Pattern of Selected Candidate Genes and MRKH Phenotypes
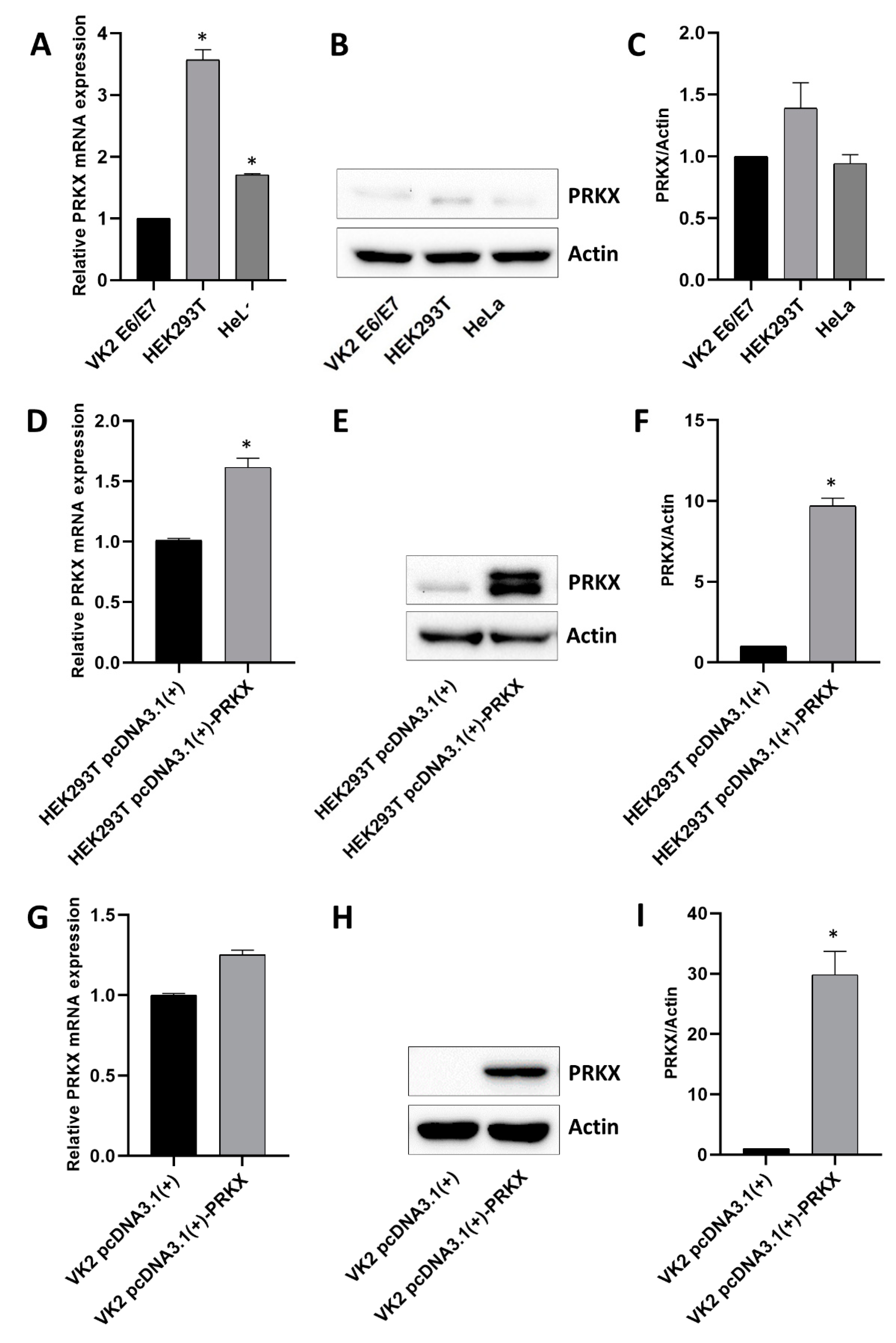
3.3. Assessment of Basal PRKX Expression Levels in Selected Human Cell Lines and PRKX Expression Vector’s Functionality
3.4. PRKX Overexpression in VK2 E6/E7 Cells Induces Cell Morphology Modifications
3.5. PRKX Overexpression Promotes VK2 E6/E7 Cells Migration as a Consequence of EMT Induction
3.6. PRKX Overexpression in VK2 E6/E7 Cells Affects HOX Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- George, F.W.; Wilson, J.D. Sex Determination and Differentiation; The Physiology of Reproduction; Knobil, E., Neil, J., Eds.; Raven Press: New York, NY, USA, 1994; Volume 1. [Google Scholar]
- Minami, N.; Iritani, A. Role of the oviduct in the development of the mouse embryo. Mol. Reprod. Dev. 1993, 36, 279–281. [Google Scholar] [CrossRef]
- Fontana, L.; Gentilin, B.; Fedele, L.; Gervasini, C.; Miozzo, M. Genetics of Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. Clin. Genet 2017, 91, 233–246. [Google Scholar] [CrossRef]
- Strübbe, E.H.; Cremers, C.W.; Willemsen, W.N.; Rolland, R.; Thijn, C.J. The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome without and with associated features: Two separate entities? Clin. Dysmorphol. 1994, 3, 192–199. [Google Scholar]
- Bean, E.J.; Mazur, T.; Robinson, A.D. Mayer-Rokitansky-Küster-Hauser syndrome: Sexuality, psychological effects, and quality of life. J. Pediatr Adolesc. Gynecol. 2009, 22, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Herlin, M.K.; Petersen, M.B.; Brannstrom, M. Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome: A comprehensive update. Orphanet. J. Rare Dis. 2020, 15, 214. [Google Scholar] [CrossRef]
- Williams, L.S.; Demir Eksi, D.; Shen, Y.; Lossie, A.C.; Chorich, L.P.; Sullivan, M.E.; Phillips, J.A., 3rd; Erman, M.; Kim, H.G.; Alper, O.M.; et al. Genetic analysis of Mayer-Rokitansky-Kuster-Hauser syndrome in a large cohort of families. Fertil Steril 2017, 108, 145–151.e2. [Google Scholar] [CrossRef] [Green Version]
- Duru, U.A.; Laufer, M.R. Discordance in Mayer-von Rokitansky-Kuster-Hauser Syndrome noted in monozygotic twins. J. Pediatr. Adolesc. Gynecol. 2009, 22, e73-5. [Google Scholar] [CrossRef]
- Lischke, J.H.; Curtis, C.H.; Lamb, E.J. Discordance of vaginal agenesis in monozygotic twins. Obstet. Gynecol. 1973, 41, 920–924. [Google Scholar]
- Regenstein, A.C.; Berkeley, A.S. Discordance of mullerian agenesis in monozygotic twins. A case report. J. Reprod. Med. 1991, 36, 396–397. [Google Scholar]
- Rall, K.; Eisenbeis, S.; Barresi, G.; Rückner, D.; Walter, M.; Poths, S.; Wallwiener, D.; Riess, O.; Bonin, M.; Brucker, S. Mayer-Rokitansky-Küster-Hauser syndrome discordance in monozygotic twins: Matrix metalloproteinase 14, low-density lipoprotein receptor-related protein 10, extracellular matrix, and neoangiogenesis genes identified as candidate genes in a tissue-specific mosaicism. Fertil. Steril. 2015, 103, 494–502.e3. [Google Scholar]
- Laronda, M.M.; Unno, K.; Ishi, K.; Serna, V.A.; Butler, L.M.; Mills, A.A.; Orvis, G.D.; Behringer, R.R.; Deng, C.; Sinha, S.; et al. Diethylstilbestrol induces vaginal adenosis by disrupting SMAD/RUNX1-mediated cell fate decision in the Mullerian duct epithelium. Dev. Biol. 2013, 381, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLachlan, J.A. Transplacental effects of diethylstilbestrol in mice. Natl. Cancer Inst. Monogr. 1979, 51, 67–72. [Google Scholar]
- Rall, K.; Barresi, G.; Walter, M.; Poths, S.; Haebig, K.; Schaeferhoff, K.; Schoenfisch, B.; Riess, O.; Wallwiener, D.; Bonin, M.; et al. A combination of transcriptome and methylation analyses reveals embryologically-relevant candidate genes in MRKH patients. Orphanet. J. Rare Dis. 2011, 6, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nodale, C.; Ceccarelli, S.; Giuliano, M.; Cammarota, M.; D’Amici, S.; Vescarelli, E.; Maffucci, D.; Bellati, F.; Panici, P.B.; Romano, F.; et al. Gene expression profile of patients with Mayer-Rokitansky-Küster-Hauser syndrome: New insights into the potential role of developmental pathways. PLoS ONE 2014, 9, e91010. [Google Scholar] [CrossRef] [Green Version]
- Hentrich, T.; Koch, A.; Weber, N.; Kilzheimer, A.; Maia, A.; Burkhardt, S.; Rall, K.; Casadei, N.; Kohlbacher, O.; Riess, O.; et al. The Endometrial Transcription Landscape of MRKH Syndrome. Front Cell Dev. Biol. 2020, 8, 572281. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Bilyk, O.; Coatham, M.; Jewer, M.; Postovit, L.M. Epithelial-to-Mesenchymal Transition in the Female Reproductive Tract: From Normal Functioning to Disease Pathology. Front Oncol. 2017, 7, 145. [Google Scholar] [CrossRef] [Green Version]
- Pontecorvi, P.; Bernardini, L.; Capalbo, A.; Ceccarelli, S.; Megiorni, F.; Vescarelli, E.; Bottillo, I.; Preziosi, N.; Fabbretti, M.; Perniola, G.; et al. Protein-protein interaction network analysis applied to DNA copy number profiling suggests new perspectives on the aetiology of Mayer-Rokitansky-Küster-Hauser syndrome. Sci. Rep. 2021, 11, 448. [Google Scholar] [CrossRef]
- Li, X.; Li, H.P.; Amsler, K.; Hyink, D.; Wilson, P.D.; Burrow, C.R. PRKX, a phylogenetically and functionally distinct cAMP-dependent protein kinase, activates renal epithelial cell migration and morphogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 9260–9565. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Li, Q.; Alberts, I.; Li, X. PRKX, a Novel cAMP-Dependent Protein Kinase Member, Plays an Important Role in Development. J. Cell Biochem. 2016, 117, 566–573. [Google Scholar] [CrossRef]
- Robboy, S.J.; Kurita, T.; Baskin, L.; Cunha, G.R. New insights into human female reproductive tract development. Differentiation 2017, 97, 9–22. [Google Scholar] [CrossRef]
- Kobayashi, A.; Behringer, R.R. Developmental genetics of the female reproductive tract in mammals. Nat. Rev. Genet. 2003, 4, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, N.; Renkema, K.Y.; Bongers, E.M.; Giles, R.H.; Knoers, N.V. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat. Rev. Nephrol. 2015, 11, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Nodale, C.; Vescarelli, E.; D’Amici, S.; Maffucci, D.; Ceccarelli, S.; Monti, M.; Benedetti Panici, P.; Romano, F.; Angeloni, A.; Marchese, C. Characterization of human vaginal mucosa cells for autologous in vitro cultured vaginal tissue transplantation in patients with MRKH syndrome. Biomed. Res. Int. 2014, 2014, 201518. [Google Scholar] [CrossRef]
- Panici, P.B.; Ruscito, I.; Gasparri, M.L.; Maffucci, D.; Marchese, C.; Bellati, F. Vaginal reconstruction with the Abbè-McIndoe technique: From dermal grafts to autologous in vitro cultured vaginal tissue transplant. Semin. Reprod Med. 2011, 29, 45–54. [Google Scholar] [CrossRef]
- Benedetti Panici, P.; Maffucci, D.; Ceccarelli, S.; Vescarelli, E.; Perniola, G.; Muzii, L.; Marchese, C. Autologous in vitro cultured vaginal tissue for vaginoplasty in women with Mayer-Rokitansky-Küster-Hauser syndrome: Anatomic and functional results. J. Minim. Invasive Gynecol. 2015, 22, 205–211. [Google Scholar] [CrossRef]
- Pontecorvi, P.; Banki, M.A.; Zampieri, C.; Zalfa, C.; Azmoon, P.; Kounnas, M.Z.; Marchese, C.; Gonias, S.L.; Mantuano, E. Fibrinolysis protease receptors promote activation of astrocytes to express pro-inflammatory cytokines. J. Neuroinflammation 2019, 16, 257. [Google Scholar] [CrossRef]
- Ringnér, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, S.; Nodale, C.; Vescarelli, E.; Pontecorvi, P.; Manganelli, V.; Casella, G.; Onesti, M.G.; Sorice, M.; Romano, F.; Angeloni, A.; et al. Neuropilin 1 Mediates Keratinocyte Growth Factor Signaling in Adipose-Derived Stem Cells: Potential Involvement in Adipogenesis. Stem Cells Int. 2018, 2018, 1075156. [Google Scholar] [CrossRef]
- Vescarelli, E.; Gerini, G.; Megiorni, F.; Anastasiadou, E.; Pontecorvi, P.; Solito, L.; De Vitis, C.; Camero, S.; Marchetti, C.; Mancini, R.; et al. MiR-200c sensitizes Olaparib-resistant ovarian cancer cells by targeting Neuropilin 1. J. Exp. Clin. Cancer Res. 2020, 39, 3. [Google Scholar] [CrossRef]
- Lotti, L.V.; Rotolo, S.; Francescangeli, F.; Frati, L.; Torrisi, M.R.; Marchese, C. AKT and MAPK signaling in KGF-treated and UVB-exposed human epidermal cells. J. Cell Physiol. 2007, 212, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Camero, S.; Ceccarelli, S.; McDowell, H.P.; Mannarino, O.; Marampon, F.; Pizer, B.; Shukla, R.; Pizzuti, A.; Marchese, C.; et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget 2016, 7, 79342–79356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Scardoni, G.; Petterlini, M.; Laudanna, C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009, 25, 2857–2859. [Google Scholar] [CrossRef] [Green Version]
- Jacquinet, A.; Boujemla, B.; Fasquelle, C.; Thiry, J.; Josse, C.; Lumaka, A.; Brischoux-Boucher, E.; Dubourg, C.; David, V.; Pasquier, L.; et al. GREB1L variants in familial and sporadic hereditary urogenital adysplasia and Mayer-Rokitansky-Kuster-Hauser syndrome. Clin. Genet. 2020, 98, 126–137. [Google Scholar] [CrossRef]
- Kaiser, H.F. An index of factorial simplicity. Psychometrika 1974, 39, 31–36. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-β1-Induced Profibrotic Signaling. Cell Physiol. Biochem. 2015, 36, 1911–1927. [Google Scholar] [CrossRef]
- Shaikh, D.; Zhou, Q.; Chen, T.; Ibe, J.C.; Raj, J.U.; Zhou, G. cAMP-dependent protein kinase is essential for hypoxia-mediated epithelial-mesenchymal transition, migration, and invasion in lung cancer cells. Cell Signal 2012, 24, 2396–2406. [Google Scholar] [CrossRef]
- Aguiari, G.; Varani, K.; Bogo, M.; Mangolini, A.; Vincenzi, F.; Durante, C.; Gessi, S.; Sacchetto, V.; Catizone, L.; Harris, P.; et al. Deficiency of polycystic kidney disease-1 gene (PKD1) expression increases A(3) adenosine receptors in human renal cells: Implications for cAMP-dependent signalling and proliferation of PKD1-mutated cystic cells. Biochim. Biophys. Acta 2009, 1792, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Schiebel, K.; Winkelmann, M.; Mertz, A.; Xu, X.; Page, D.C.; Weil, D.; Petit, C.; Rappold, G.A. Abnormal XY interchange between a novel isolated protein kinase gene, PRKY, and its homologue, PRKX, accounts for one third of all (Y+)XX males and (Y-)XY females. Hum. Mol. Genet. 1997, 6, 1985–1989. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, B.; Chiorini, J.A.; Ma, Y.; Kotin, R.M.; Herberg, F.W. PrKX is a novel catalytic subunit of the cAMP-dependent protein kinase regulated by the regulatory subunit type I. J. Biol. Chem. 1999, 274, 5370–5378. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hyink, D.P.; Polgar, K.; Gusella, G.L.; Wilson, P.D.; Burrow, C.R. Protein kinase X activates ureteric bud branching morphogenesis in developing mouse metanephric kidney. J. Am. Soc. Nephrol. 2005, 16, 3543–3552. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hyink, D.P.; Radbill, B.; Sudol, M.; Zhang, H.; Zheleznova, N.N.; Wilson, P.D. Protein kinase-X interacts with Pin-1 and Polycystin-1 during mouse kidney development. Kidney Int. 2009, 76, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Silverman, E.K.; Schmidt, H.H.H.W.; Anastasiadou, E.; Altucci, L.; Angelini, M.; Badimon, L.; Balligand, J.L.; Benincasa, G.; Capasso, G.; Conte, F.; et al. Molecular networks in Network Medicine: Development and applications. Wiley Interdiscip Rev. Syst. Biol. Med. 2020, 12, e1489. [Google Scholar] [CrossRef]
- Hossain, A.; Saunders, G.F. Synergistic cooperation between the beta-catenin signaling pathway and steroidogenic factor 1 in the activation of the Mullerian inhibiting substance type II receptor. J. Biol. Chem. 2003, 278, 26511–26516. [Google Scholar] [CrossRef] [Green Version]
- Allard, S.; Adin, P.; Gouedard, L.; di Clemente, N.; Josso, N.; Orgebin-Crist, M.C.; Picard, J.Y.; Xavier, F. Molecular mechanisms of hormone-mediated Mullerian duct regression: Involvement of beta-catenin. Development 2000, 127, 3349–3360. [Google Scholar] [CrossRef]
- Huang, L.; Ren, J.; Chen, D.; Li, Y.; Kharbanda, S.; Kufe, D. MUC1 cytoplasmic domain coactivates Wnt target gene transcription and confers transformation. Cancer Biol. Ther. 2003, 2, 702–706. [Google Scholar] [CrossRef]
- Juan, A.H.; Lei, H.; Bhargava, P.; Lebrun, M.; Ruddle, F.H. Multiple roles of hoxc8 in skeletal development. Ann. N. Y. Acad. Sci. 2006, 1068, 87–94. [Google Scholar] [CrossRef]
- Kwon, Y.; Shin, J.; Park, H.W.; Kim, M.H. Dynamic expression pattern of Hoxc8 during mouse early embryogenesis. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2005, 283, 187–192. [Google Scholar] [CrossRef]
- Brophy, P.D.; Rasmussen, M.; Parida, M.; Bonde, G.; Darbro, B.W.; Hong, X.; Clarke, J.C.; Peterson, K.A.; Denegre, J.; Schneider, M.; et al. A Gene Implicated in Activation of Retinoic Acid Receptor Targets Is a Novel Renal Agenesis Gene in Humans. Genetics 2017, 207, 215–228. [Google Scholar] [CrossRef]
- De Tomasi, L.; David, P.; Humbert, C.; Silbermann, F.; Arrondel, C.; Tores, F.; Fouquet, S.; Desgrange, A.; Niel, O.; Bole-Feysot, C.; et al. Mutations in GREB1L Cause Bilateral Kidney Agenesis in Humans and Mice. Am. J. Hum. Genet 2017, 101, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Sanna-Cherchi, S.; Khan, K.; Westland, R.; Krithivasan, P.; Fievet, L.; Rasouly, H.M.; Ionita-Laza, I.; Capone, V.P.; Fasel, D.A.; Kiryluk, K.; et al. Exome-wide Association Study Identifies GREB1L Mutations in Congenital Kidney Malformations. Am. J. Hum. Genet 2017, 101, 1034. [Google Scholar] [CrossRef] [Green Version]
- Herlin, M.K.; Le, V.Q.; Højland, A.T.; Ernst, A.; Okkels, H.; Petersen, A.C.; Petersen, M.B.; Pedersen, I.S. Whole-exome sequencing identifies a GREB1L variant in a three-generation family with Müllerian and renal agenesis: A novel candidate gene in Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome. A case report. Hum. Reprod 2019, 34, 1838–1846. [Google Scholar] [CrossRef]
- Rosselot, C.; Spraggon, L.; Chia, I.; Batourina, E.; Riccio, P.; Lu, B.; Niederreither, K.; Dolle, P.; Duester, G.; Chambon, P.; et al. Non-cell-autonomous retinoid signaling is crucial for renal development. Development 2010, 137, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Vivante, A.; Mann, N.; Yonath, H.; Weiss, A.C.; Getwan, M.; Kaminski, M.M.; Bohnenpoll, T.; Teyssier, C.; Chen, J.; Shril, S.; et al. A Dominant Mutation in Nuclear Receptor Interacting Protein 1 Causes Urinary Tract Malformations via Dysregulation of Retinoic Acid Signaling. J. Am. Soc. Nephrol. 2017, 28, 2364–2376. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Sato, T.; Iguchi, T.; Takasugi, N. Retinoic acid signaling determines the fate of the uterus from the mouse Mullerian duct. Reprod. Toxicol. 2019, 86, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Panici, P.B.; Bellati, F.; Boni, T.; Francescangeli, F.; Frati, L.; Marchese, C. Vaginoplasty using autologous in vitro cultured vaginal tissue in a patient with Mayer-von-Rokitansky-Kuster-Hauser syndrome. Hum. Reprod 2007, 22, 2025–2028. [Google Scholar] [CrossRef] [Green Version]
- Sabatucci, I.; Palaia, I.; Marchese, C.; Muzii, L.; Morte, C.D.; Giorgini, M.; Musella, A.; Ceccarelli, S.; Vescarelli, E.; Panici, P.B. Treatment of the Mayer-Rokitansky-Küster-Hauser syndrome with autologous in vitro cultured vaginal tissue: Descriptive study of long-term results and patient outcomes. BJOG 2019, 126, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessy, L.A.; Mazzocchi, M.; Corrias, F.; Ceccarelli, S.; Marchese, C.; Scuderi, N. The use of cultured autologous oral epithelial cells for vaginoplasty in male-to-female transsexuals: A feasibility, safety, and advantageousness clinical pilot study. Plast. Reconstr. Surg. 2014, 133, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Onesti, M.G.; Carella, S.; Ceccarelli, S.; Marchese, C.; Scuderi, N. The Use of Human Adipose-Derived Stem Cells in the Treatment of Physiological and Pathological Vulvar Dystrophies. Stem. Cells Int. 2016, 2561461. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, P.; Trerotola, M.; Franck, J.; Cardon, T.; Marchisio, M.; Fournier, I.; Salzet, M.; Maffia, M.; Vergara, D. The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Biol. 2019, 58, 1–10. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [Green Version]
- Plygawko, A.T.; Kan, S.; Campbell, K. Epithelial-mesenchymal plasticity: Emerging parallels between tissue morphogenesis and cancer metastasis. Philos. Trans R Soc. Lond B Biol. Sci. 2020, 375, 20200087. [Google Scholar] [CrossRef]
- Giacomelli, C.; Daniele, S.; Romei, C.; Tavanti, L.; Neri, T.; Piano, I.; Celi, A.; Martini, C.; Trincavelli, M.L. The A2B Adenosine Receptor Modulates the Epithelial- Mesenchymal Transition through the Balance of cAMP/PKA and MAPK/ERK Pathway Activation in Human Epithelial Lung Cells. Front Pharmacol. 2018, 9, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.S.J.; So, M.; Tang, C.S.M.; Karim, A.; Porsch, R.M.; Wong, C.; Yu, M.; Yeung, F.; Xia, H.; Zhang, R.; et al. De novo mutations in Caudal Type Homeo Box transcription Factor 2 (CDX2) in patients with persistent cloaca. Hum. Mol. Genet 2018, 27, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Moisan, V.; Bomgardner, D.; Tremblay, J.J. Expression of the Ladybird-like homeobox 2 transcription factor in the developing mouse testis and epididymis. BMC Dev. Biol. 2008, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Hallonet, M.; Hollemann, T.; Wehr, R.; Jenkins, N.A.; Copeland, N.G.; Pieler, T.; Gruss, P. Vax1 is a novel homeobox-containing gene expressed in the developing anterior ventral forebrain. Development 1998, 125, 2599–2610. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Yoshida, M.; Kuratani, S.; Matsuo, I.; Aizawa, S. Defects of urogenital development in mice lacking Emx2. Development 1997, 124, 1653–1664. [Google Scholar] [CrossRef]
- Debeer, P.; Bacchelli, C.; Scambler, P.J.; De Smet, L.; Fryns, J.P.; Goodman, F.R. Severe digital abnormalities in a patient heterozygous for both a novel missense mutation in HOXD13 and a polyalanine tract expansion in HOXA13. J. Med. Genet 2002, 39, 852–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmita, M.; Kondo, T.; Duboule, D. Targeted inversion of a polar silencer within the HoxD complex re-allocates domains of enhancer sharing. Nat. Genet 2000, 26, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, S.; Johnson, K.R.; Yamamoto, M.; Kuroiwa, A.; Duboule, D. The mouse Hoxd13(spdh) mutation, a polyalanine expansion similar to human type II synpolydactyly (SPD), disrupts the function but not the expression of other Hoxd genes. Dev. Biol. 2001, 237, 345–353. [Google Scholar] [CrossRef]
- Patterson, L.T.; Pembaur, M.; Potter, S.S. Hoxa11 and Hoxd11 regulate branching morphogenesis of the ureteric bud in the developing kidney. Development 2001, 128, 2153–2161. [Google Scholar] [CrossRef]
- Di-Poi, N.; Zakany, J.; Duboule, D. Distinct roles and regulations for HoxD genes in metanephric kidney development. PLoS Genet. 2007, 3, e232. [Google Scholar] [CrossRef] [Green Version]
- Gervasini, C.; Grati, F.R.; Lalatta, F.; Tabano, S.; Gentilin, B.; Colapietro, P.; De Toffol, S.; Frontino, G.; Motta, F.; Maitz, S.; et al. SHOX duplications found in some cases with type I Mayer-Rokitansky-Kuster-Hauser syndrome. Genet. Med. 2010, 12, 634–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, M.; Sun, Y.; Yang, H.L.; Zhang, B.; Wen, J.; Shi, B.K. DLX1, a binding protein of beta-catenin, promoted the growth and migration of prostate cancer cells. Exp. Cell Res. 2018, 363, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Lewis, R.A.; Yu, L.; He, F.; Liu, H.; Tang, R.; Shi, J.; Sun, X.; Martin, J.F.; Wang, D.; Yang, J.; et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev. Biol. 2009, 327, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Self, M.; Lagutin, O.V.; Bowling, B.; Hendrix, J.; Cai, Y.; Dressler, G.R.; Oliver, G. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 2006, 25, 5214–5228. [Google Scholar] [CrossRef]
- Lenti, E.; Farinello, D.; Yokoyama, K.K.; Penkov, D.; Castagnaro, L.; Lavorgna, G.; Wuputra, K.; Sandell, L.L.; Tjaden, N.E.; Bernassola, F.; et al. Transcription factor TLX1 controls retinoic acid signaling to ensure spleen development. J. Clin. Investig. 2016, 126, 2452–2464. [Google Scholar] [CrossRef] [Green Version]
- Di Pasquale, G.; Stacey, S.N. Adeno-associated virus Rep78 protein interacts with protein kinase A and its homolog PRKX and inhibits CREB-dependent transcriptional activation. J. Virol. 1998, 72, 7916–7925. [Google Scholar] [CrossRef] [Green Version]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster 1 (Purple) | Cluster 2 (Green) | Cluster 3 (Yellow) | |||
|---|---|---|---|---|---|
| Patients | MRKH Type | Patients | MRKH Type | Patients | MRKH Type |
| 8 | I | 7 | I | 25 | II |
| 10 | II | 9 | I | 26 | I |
| 12 | II | 11 | I | 27 | I |
| 13 | II | 14 | II | 39 | II |
| 15 | I | 19 | II | 40 | I |
| 16 | I | 20 | II | ||
| 17 | I | 22 | II | ||
| 18 | II | 29 | II | ||
| 21 | I | 31 | II | ||
| 23 | I | 33 | II | ||
| 24 | II | 35 | I | ||
| 28 | I | ||||
| 32 | I | ||||
| 34 | II | ||||
| 36 | I | ||||
| 37 | I | ||||
| 41 | I | ||||
| 42 | I | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pontecorvi, P.; Megiorni, F.; Camero, S.; Ceccarelli, S.; Bernardini, L.; Capalbo, A.; Anastasiadou, E.; Gerini, G.; Messina, E.; Perniola, G.; et al. Altered Expression of Candidate Genes in Mayer–Rokitansky–Küster–Hauser Syndrome May Influence Vaginal Keratinocytes Biology: A Focus on Protein Kinase X. Biology 2021, 10, 450. https://doi.org/10.3390/biology10060450
Pontecorvi P, Megiorni F, Camero S, Ceccarelli S, Bernardini L, Capalbo A, Anastasiadou E, Gerini G, Messina E, Perniola G, et al. Altered Expression of Candidate Genes in Mayer–Rokitansky–Küster–Hauser Syndrome May Influence Vaginal Keratinocytes Biology: A Focus on Protein Kinase X. Biology. 2021; 10(6):450. https://doi.org/10.3390/biology10060450
Chicago/Turabian StylePontecorvi, Paola, Francesca Megiorni, Simona Camero, Simona Ceccarelli, Laura Bernardini, Anna Capalbo, Eleni Anastasiadou, Giulia Gerini, Elena Messina, Giorgia Perniola, and et al. 2021. "Altered Expression of Candidate Genes in Mayer–Rokitansky–Küster–Hauser Syndrome May Influence Vaginal Keratinocytes Biology: A Focus on Protein Kinase X" Biology 10, no. 6: 450. https://doi.org/10.3390/biology10060450
APA StylePontecorvi, P., Megiorni, F., Camero, S., Ceccarelli, S., Bernardini, L., Capalbo, A., Anastasiadou, E., Gerini, G., Messina, E., Perniola, G., Benedetti Panici, P., Grammatico, P., Pizzuti, A., & Marchese, C. (2021). Altered Expression of Candidate Genes in Mayer–Rokitansky–Küster–Hauser Syndrome May Influence Vaginal Keratinocytes Biology: A Focus on Protein Kinase X. Biology, 10(6), 450. https://doi.org/10.3390/biology10060450