
Novel Severe Hemophilia A Mouse Model with Factor VIII Intron 22 Inversion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. sgRNA Preparation
2.2. Mutant Mouse Generation
2.3. ELISA and Chromogenic Assay
2.4. Prothrombin Time and Activated Partial Thromboplastin Time Test
2.5. In Vivo Bleeding Test
2.6. Complete Blood Count
2.7. Calibrated Automated Thrombogram
2.8. Survival Rate Analysis
2.9. Histological Examination
2.10. Statistical Analysis
3. Results
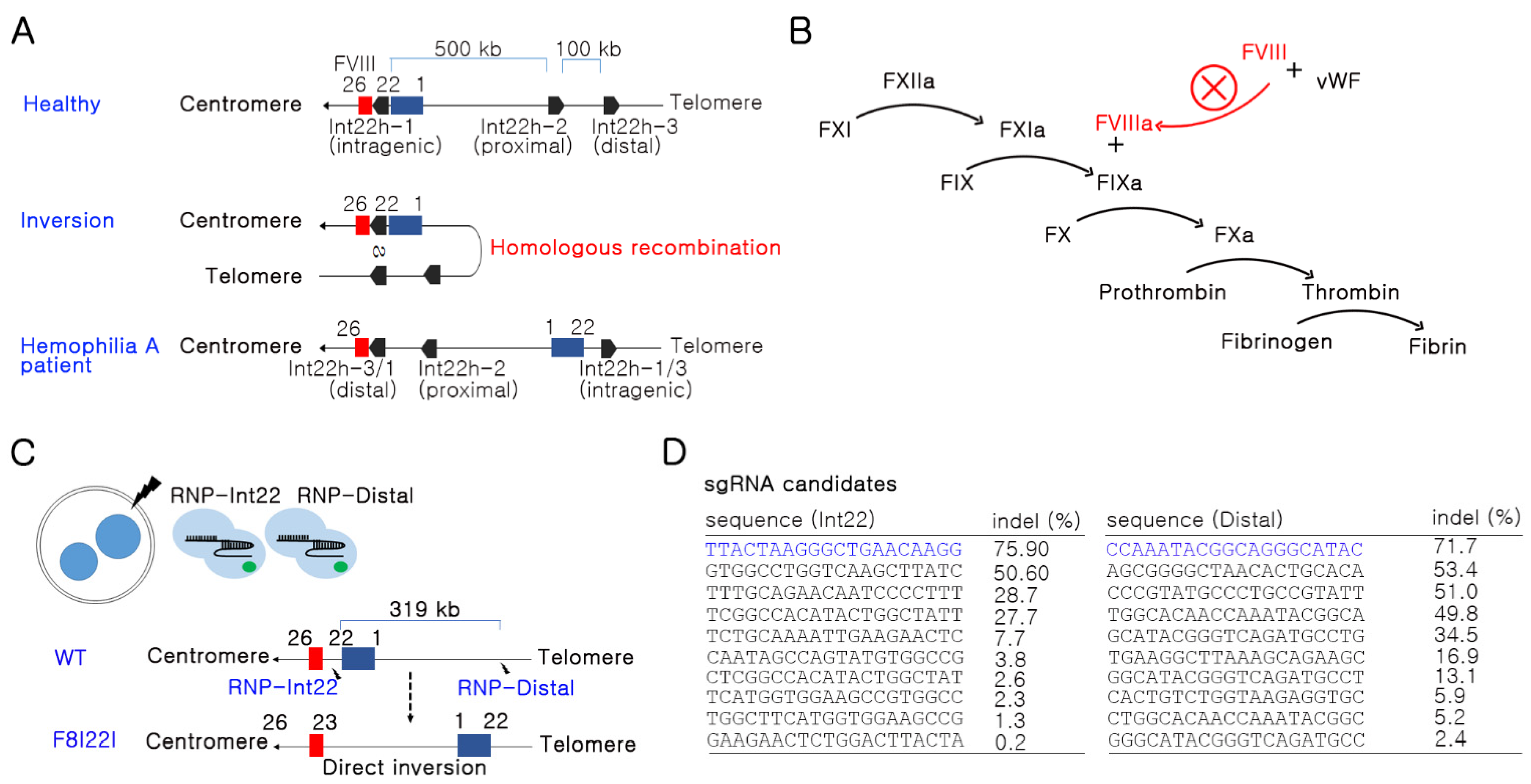
3.1. Potent sgRNA Selection in the F8 Gene by In Vitro Screening
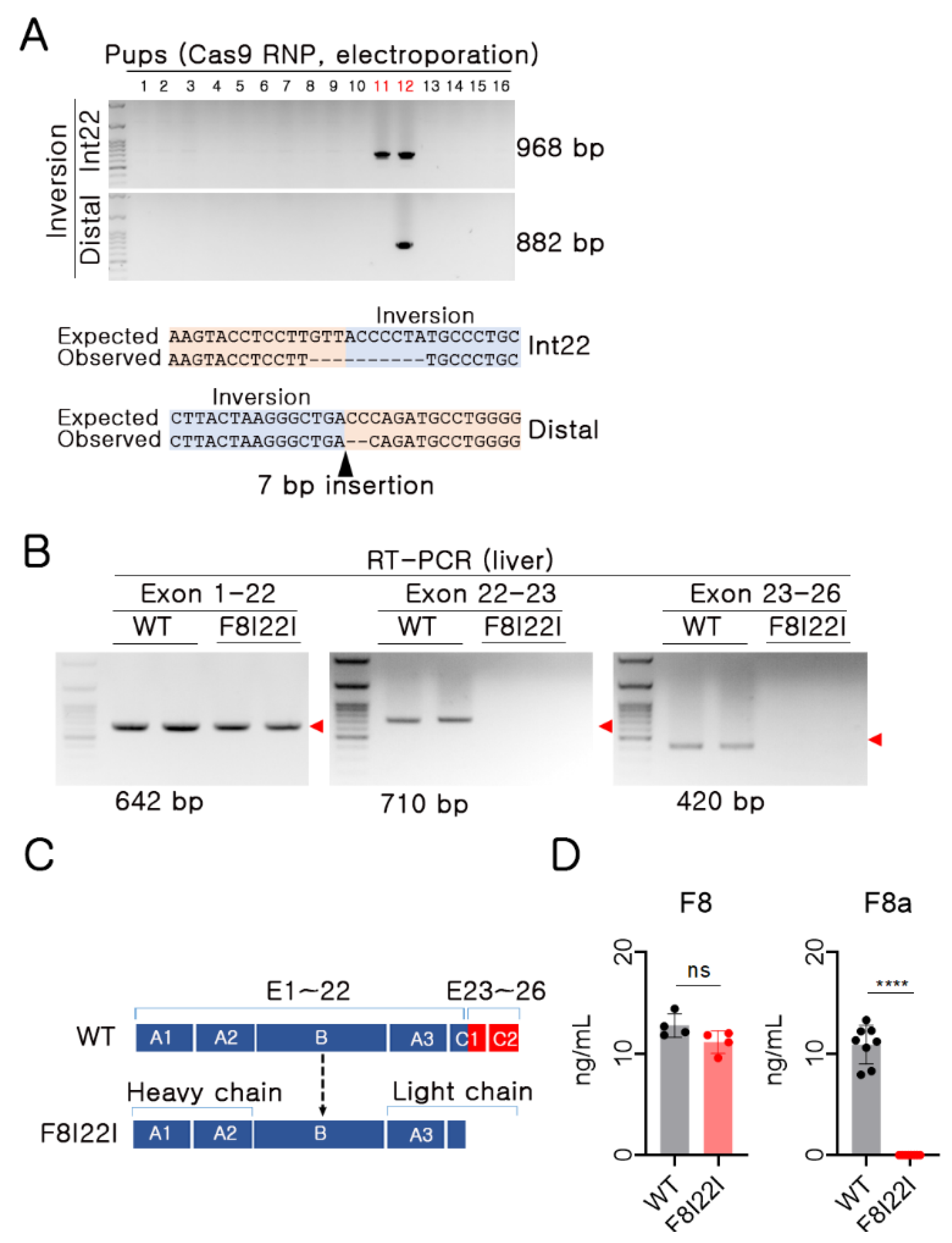
3.2. Functional F8 Deficiency in F8I22I Mouse
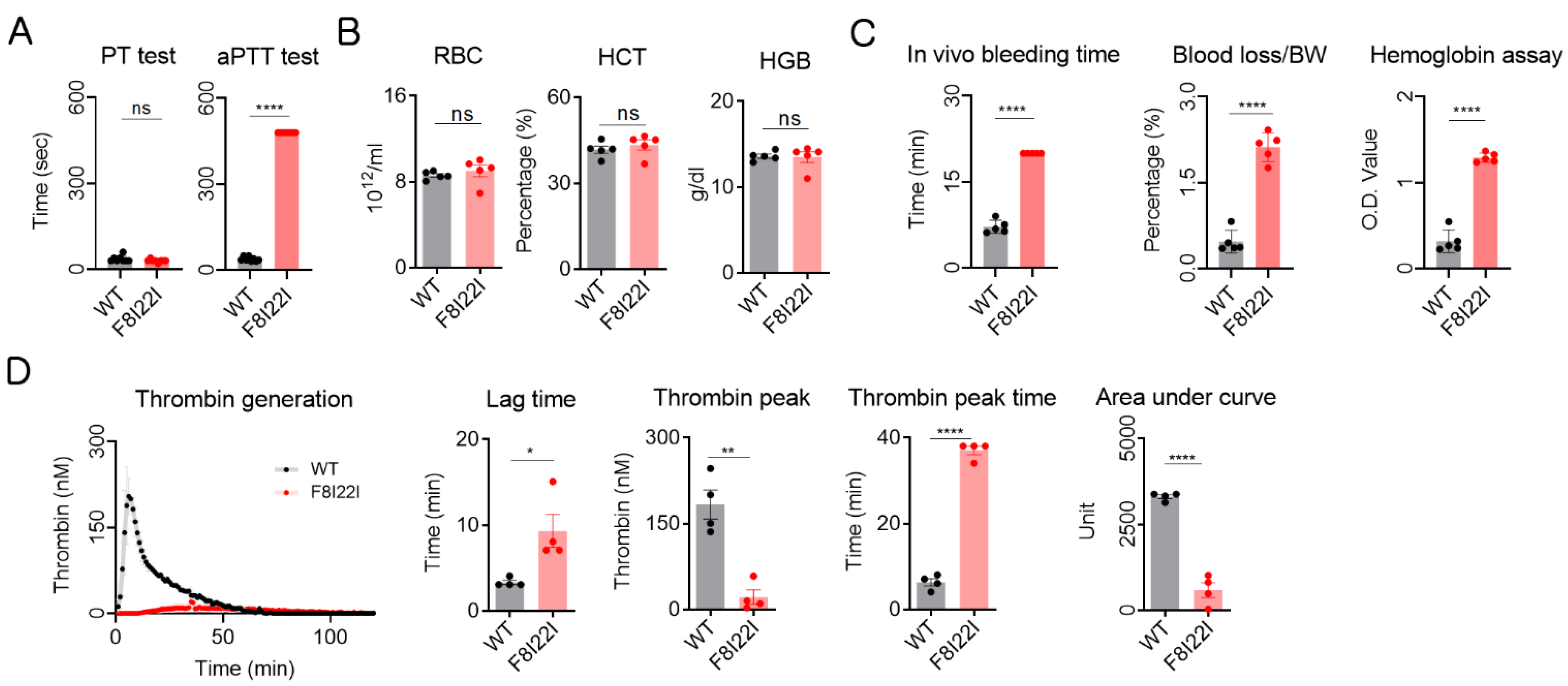
3.3. Laboratory Diagnosis for Hemophilia
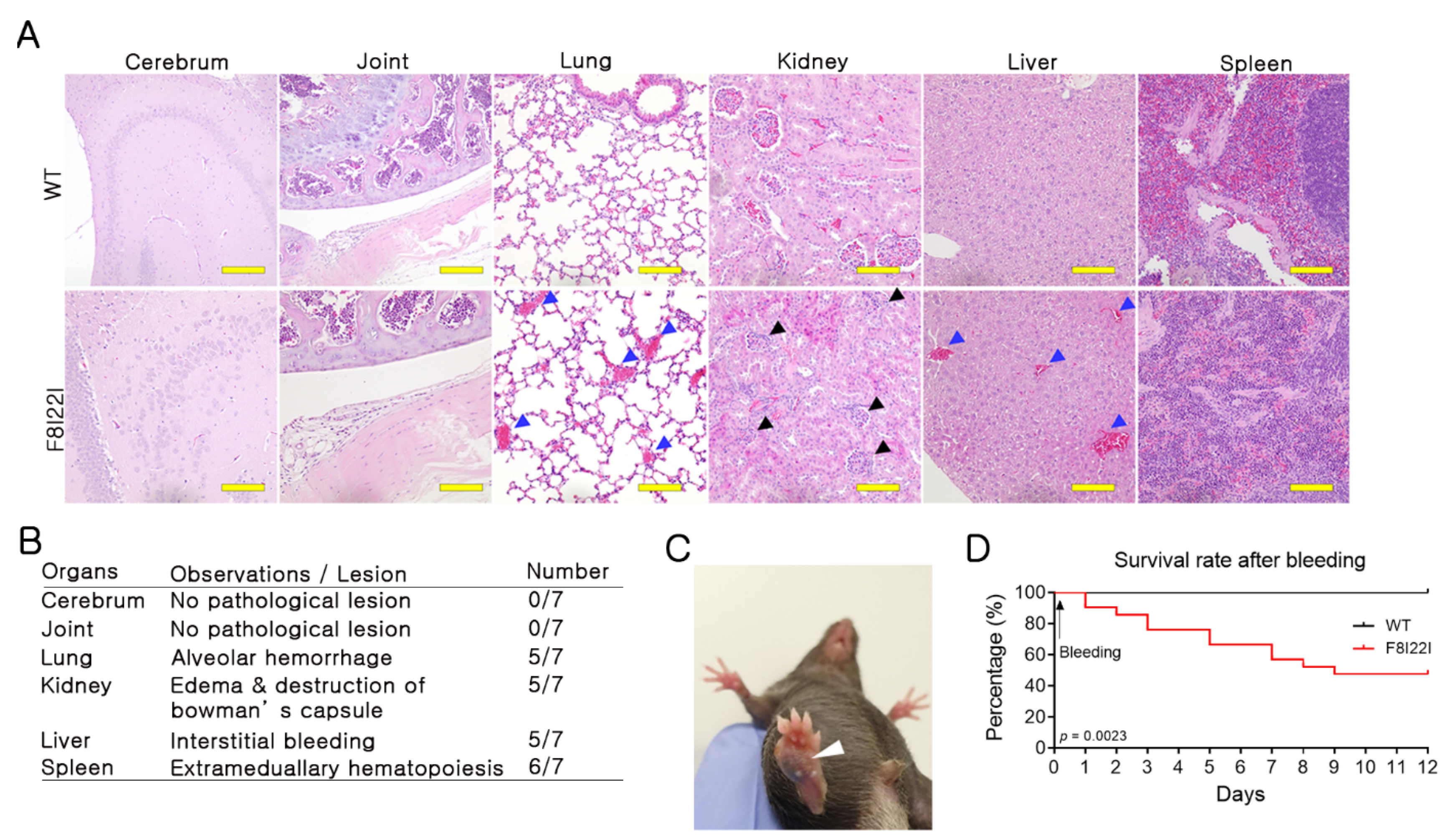
3.4. Bleeding in Lung and Liver of F8I22I Mouse
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jamil, M.A.; Sharma, A.; Nuesgen, N.; Pezeshkpoor, B.; Heimbach, A.; Pavlova, A.; Oldenburg, J.; El-Maarri, O. F8 Inversions at Xq28 Causing Hemophilia A Are Associated With Specific Methylation Changes: Implication for Molecular Epigenetic Diagnosis. Front Genet. 2019, 10, 508. [Google Scholar] [CrossRef]
- Bowen, D.J. Haemophilia A and haemophilia B: Molecular insights. Mol. Pathol. 2002, 55, 127–144. [Google Scholar] [CrossRef] [Green Version]
- Graw, J.; Brackmann, H.H.; Oldenburg, J.; Schneppenheim, R.; Spannagl, M.; Schwaab, R. Haemophilia A: From mutation analysis to new therapies. Nat. Rev. Genet. 2005, 6, 488–501. [Google Scholar] [CrossRef]
- Gouw, S.C.; van den Berg, H.M.; Oldenburg, J.; Astermark, J.; de Groot, P.G.; Margaglione, M.; Thompson, A.R.; van Heerde, W.; Boekhorst, J.; Miller, C.H.; et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: Systematic review and meta-analysis. Blood 2012, 119, 2922–2934. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, J.M.; Fletcher, S.N.; Huston, H.; Roberge, S.; Martin, B.K.; Kircher, M.; Josephson, N.C.; Shendure, J.; Ruuska, S.; Koerper, M.A.; et al. Novel approach to genetic analysis and results in 3000 hemophilia patients enrolled in the My Life, Our Future initiative. Blood Adv. 2017, 1, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, A.; Brewer, A.K.; Mauser-Bunschoten, E.P.; Key, N.S.; Kitchen, S.; Llinas, A.; Ludlam, C.A.; Mahlangu, J.N.; Mulder, K.; Poon, M.C.; et al. Guidelines for the management of hemophilia. Haemophilia 2013, 19, e1-47. [Google Scholar] [CrossRef] [PubMed]
- Evatt, B.L.; Robillard, L. Establishing haemophilia care in developing countries: Using data to overcome the barrier of pessimism. Haemophilia 2000, 6, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; Van Der Bom, J.G.; Van Den Berg, H.M.; Zewald, R.A.; Ploos Van Amstel, J.K.; Mauser-Bunschoten, E.P. Influence of the type of F8 gene mutation on inhibitor development in a single centre cohort of severe haemophilia A patients. Haemophilia 2011, 17, 275–281. [Google Scholar] [CrossRef]
- Peters, R.; Harris, T. Advances and innovations in haemophilia treatment. Nat. Rev. Drug Discov. 2018, 17, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.T.; Fan, M.N.; Yang, Y.L.; Chou, S.C.; Yu, I.S.; Lin, S.W. Current animal models of hemophilia: The state of the art. Thromb J. 2016, 14, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozier, J.N.; Dutra, A.; Pak, E.; Zhou, N.; Zheng, Z.; Nichols, T.C.; Bellinger, D.A.; Read, M.; Morgan, R.A. The Chapel Hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc. Natl. Acad. Sci. USA 2002, 99, 12991–12996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hough, C.; Kamisue, S.; Cameron, C.; Notley, C.; Tinlin, S.; Giles, A.; Lillicrap, D. Aberrant splicing and premature termination of transcription of the FVIII gene as a cause of severe canine hemophilia A: Similarities with the intron 22 inversion mutation in human hemophilia. Thromb. Haemost. 2002, 87, 659–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hattab, A.W.; Schaaf, C.P.; Fang, P.; Roeder, E.; Kimonis, V.E.; Church, J.A.; Patel, A.; Cheung, S.W. Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: New cases and literature review. BMC Med. Genet. 2015, 16, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shou, J.; Guo, Y.; Tang, Y.; Wu, Y.; Jia, Z.; Zhai, Y.; Chen, Z.; Xu, Q.; Wu, Q. Efficient inversions and duplications of mammalian regulatory DNA elements and gene clusters by CRISPR/Cas9. J. Mol. Cell Biol. 2015, 7, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lim, K.; Kim, J.S.; Bae, S. Cas-analyzer: An online tool for assessing genome editing results using NGS data. Bioinformatics 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Jennings, N.L.; Dart, A.M.; Du, X.J. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J. Exp. Med. 2012, 2, 30–36. [Google Scholar] [CrossRef]
- Hegde, A.; Nair, R.; Upadhyaya, S. Spontaneous intracerebral hemorrhage in hemophiliacs-A treatment dilemma. Int. J. Surg. Case Rep. 2016, 29, 17–19. [Google Scholar] [CrossRef] [Green Version]
- Esposito, P.; Rampino, T.; Gregorini, M.; Fasoli, G.; Gamba, G.; Dal Canton, A. Renal diseases in haemophilic patients: Pathogenesis and clinical management. Eur. J. Haematol. 2013, 91, 287–294. [Google Scholar] [CrossRef]
- Mahan, M.E.; Jordan, R.M.; Me, J.C.P.; Toy, F. A rare case of spontaneous splenic rupture complicated by hemophilia A. J. Surg. Case Rep. 2019, 2019, rjz259. [Google Scholar] [CrossRef] [PubMed]
- Knobe, K.; Berntorp, E. Haemophilia and joint disease: Pathophysiology, evaluation, and management. J. Comorb. 2011, 1, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Kweon, J.; Kim, E.; Kim, S.; Kim, J.S. Targeted chromosomal duplications and inversions in the human genome using zinc finger nucleases. Genome. Res. 2012, 22, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, L.; Lawler, A.M.; Antonarakis, S.E.; High, K.A.; Gearhart, J.D.; Kazazian, H.H., Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat. Genet. 1995, 10, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.I.; Muzyczka, N. AAV: An Overview of Unanswered Questions. Hum. Gene. Ther. 2017, 28, 308–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doshi, B.S.; Arruda, V.R. Gene therapy for hemophilia: What does the future hold? Ther. Adv. Hematol. 2018, 9, 273–293. [Google Scholar] [CrossRef]
- Patel, S.R.; Lundgren, T.S.; Spencer, H.T.; Doering, C.B. The Immune Response to the fVIII Gene Therapy in Preclinical Models. Front. Immunol. 2020, 11, 494. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.H.; Kim, D.W.; Kim, J.S. Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Kim, J.; Kweon, J.; Son, J.S.; Lee, J.S.; Yoo, J.E.; Cho, S.R.; Kim, J.H.; Kim, J.S.; Kim, D.W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. USA 2014, 111, 9253–9258. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Hu, Z.; Li, Z.; Pang, J.; Feng, M.; Hu, X.; Wang, X.; Lin-Peng, S.; Liu, B.; Chen, F.; et al. In situ genetic correction of F8 intron 22 inversion in hemophilia A patient-specific iPSCs. Sci. Rep. 2016, 6, 18865. [Google Scholar] [CrossRef]
- Hamilton, M.; French, W.; Rhymes, N.; Collins, P. Liver haemorrhage in haemophilia--a case report and review of the literature. Haemophilia 2006, 12, 441–443. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.P.; Song, D.W.; Lee, J.H.; Lee, G.S.; Yeom, S.C. Novel Severe Hemophilia A Mouse Model with Factor VIII Intron 22 Inversion. Biology 2021, 10, 704. https://doi.org/10.3390/biology10080704
Han JP, Song DW, Lee JH, Lee GS, Yeom SC. Novel Severe Hemophilia A Mouse Model with Factor VIII Intron 22 Inversion. Biology. 2021; 10(8):704. https://doi.org/10.3390/biology10080704
Chicago/Turabian StyleHan, Jeong Pil, Dong Woo Song, Jeong Hyeon Lee, Geon Seong Lee, and Su Cheong Yeom. 2021. "Novel Severe Hemophilia A Mouse Model with Factor VIII Intron 22 Inversion" Biology 10, no. 8: 704. https://doi.org/10.3390/biology10080704