
PPAR Ligands Induce Antiviral Effects Targeting Perturbed Lipid Metabolism during SARS-CoV-2, HCV, and HCMV Infection




Abstract
:Simple Summary
Abstract
1. Introduction: Targeting Metabolism during Viral Infections
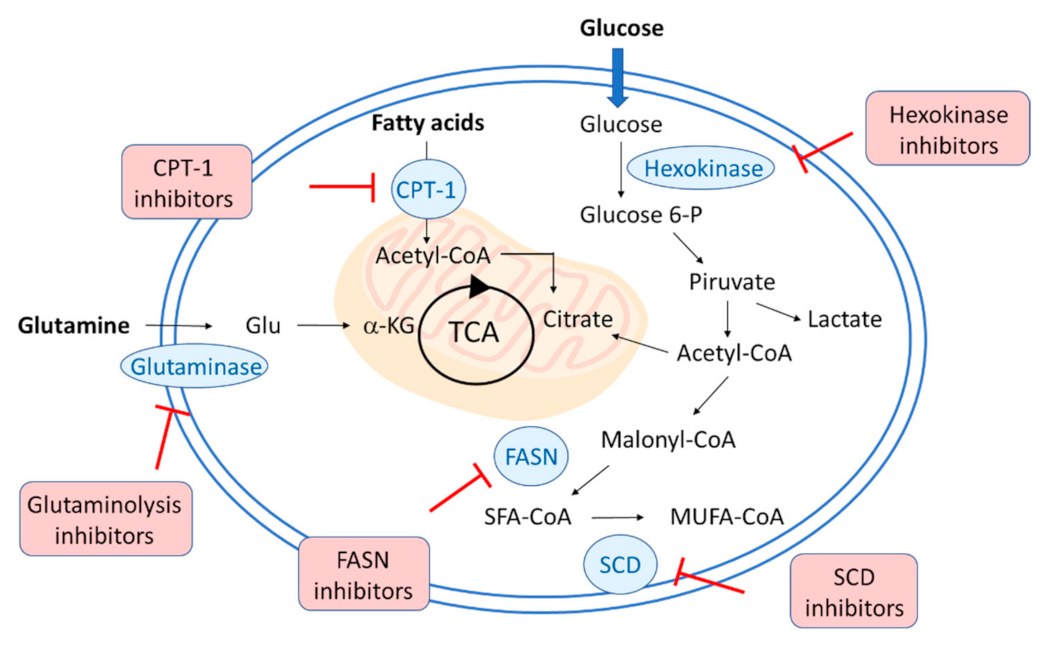
2. Changes in Lipid Metabolism during SARS-CoV2, HCV, and HCMV Infection
3. PPARs in Viral Infections
3.1. Antiviral Effects Played by PPAR Ligands in SARS-CoV-2 Infection
3.2. Antiviral Effects Played by PPAR Ligands in HCV Infection
3.3. Antiviral Effects Played by PPAR Ligands in HCMV Infection
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 13-HODE | 13-Hydroxyoctadecadienoic acid |
| 15-HETE | 15-Hydroxyeicosatetraenoic acid |
| 2-DG | 2-Deoxy-D-glucose |
| 3CL-Pro | 3-Chymotrypsin-like protease |
| 9-HODE | 9-Hydroxyoctadecadienoic acid |
| ACC | Acetyl-CoA carboxylase |
| CPT1 | Carnitine palmitoyl transferase 1 |
| DENV | Dengue virus |
| EBV | Epstein–Barr virus |
| FAO | Fatty acid oxidation |
| FAS (or FASN) | Fatty acid synthase |
| GSH | Glutathione |
| HBV | Hepatitis B virus |
| HCMV | Human cytomegalovirus |
| HCV | Hepatitis C virus |
| HIPEC | Human immortalized extravillous cytotrophoblasts |
| HIV | Human immunodeficiency virus |
| HNSCC | Head and neck cancer squamous cell carcinoma |
| HSV | Herpes simplex virus |
| IL-1β | Interleukin 1 beta |
| IL-6 | Interleukin 6 |
| JNK | c-Jun amino-terminal kinases |
| LDL | Low-density lipoprotein |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MOI | Multiplicity of infection |
| MUFAs | Monounsaturated fatty acids |
| NASH | Nonalcoholic steatohepatitis |
| NF-κB | Nuclear factor-κB |
| PEA | Palmitoylethanolamide |
| Peg-IFNα | Pegylated interferon alpha |
| PPARs | Peroxisome Proliferator-Activated Receptors |
| pUL37x1 | UL37x1 protein |
| ROS | Reactive oxygen species |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SCD | Stearoyl-CoA desaturase |
| SREBP1c | Sterol regulatory element-binding protein 1c |
| TCA | Tricarboxylic acid |
| TNF-α | Tumor necrosis factor alpha |
| VLCFAs | Very long chain fatty acids |
| VLDL | Very-low-density lipoprotein |
| NSCs | Neural stem cells |
References
- Schelhaas, M. Come in and take your coat off—How host cells provide endocytosis for virus entry. Cell Microbiol. 2010, 12, 1378–1388. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Poudevigne, E.; Effantin, G.; Bassereau, P. How to get out: ssRNA enveloped viruses and membrane fission. Curr. Opin. Virol. 2013, 3, 159–167. [Google Scholar] [CrossRef]
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front. Cell Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular metabolism is a major determinant of HIV-1 reservoir seeding in CD4+ T cells and offers an opportunity to tackle infection. Cell Metab. 2019, 29, 611–626.e5. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, M.M.; Ratcliff, A.N.; Bhat, M.; Alwarawrah, Y.; Hughes, P.; Arcos, J.; Loiselle, D.; Torrelles, J.B.; Funderburg, N.T.; Haystead, T.A.; et al. Cellular fatty acid synthase is required for late stages of HIV-1 replication. Retrovirology 2017, 14, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwerbrock, N.M.; Karlsson, E.A.; Shi, Q.; Sheridan, P.A.; Beck, M.A. Fish oil-fed mice have impaired resistance to influenza infection. J. Nutr. 2009, 139, 1588–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumbria, D.; Berber, E.; Rouse, B.T. Supplementing the diet with sodium propionate suppresses the severity of viral immuno-inflammatory lesions. J. Virol. 2021, 95, e02056-20. [Google Scholar] [CrossRef] [PubMed]
- Uyangaa, E.; Lee, H.K.; Eo, S.K. Glutamine and leucine provide enhanced protective immunity against mucosal infection with herpes simplex virus type 1. Immune Netw. 2012, 12, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Hoshino, Y.; Dowdell, K.; Bosch-Marce, M.; Myers, T.G.; Sarmiento, M.; Pesnicak, L.; Krause, P.R.; Cohen, J.I. Glutamine supplementation suppresses herpes simplex virus reactivation. J. Clin. Investig. 2017, 127, 2626–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Kurtoglu, M.; Lampidis, T.J. The wonders of 2-deoxy-D-glucose. IUBMB Life 2014, 66, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-glucose and its analogs: From diagnostic to therapeutic agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef] [Green Version]
- Courtney, R.J.; Steiner, S.M.; Benyesh-Melnick, M. Effects of 2-deoxy-D-glucose on herpes simplex virus replication. Virology 1973, 52, 447–455. [Google Scholar] [CrossRef]
- Kilbourne, E.D. Inhibition of influenza virus multiplication with a glucose antimetabolite (2-deoxy-D-glucose). Nature 1959, 183, 271–272. [Google Scholar] [CrossRef]
- Radsak, K.D.; Weder, D. Effect of 2-deoxy-D-glucose on cytomegalovirus-induced DNA synthesis in human fibroblasts. J. Gen. Virol. 1981, 57 Pt 1, 33–42. [Google Scholar] [CrossRef]
- Gualdoni, G.A.; Mayer, K.A.; Kapsch, A.M.; Kreuzberg, K.; Puck, A.; Kienzl, P.; Oberndorfer, F.; Frühwirth, K.; Winkler, S.; Blaas, D.; et al. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc. Natl. Acad. Sci. USA 2018, 115, E7158–E7165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Adhikary, A.; Woloschak, G.; Dwarakanath, B.S.; Papineni, R.V.L. A combinatorial approach of a polypharmacological adjuvant 2-deoxy-D-glucose with low dose radiation therapy to quell the cytokine storm in COVID-19 management. Int. J. Radiat. Biol. 2020, 96, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2009, 50 (Suppl. 1), S138–S143. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef]
- Flavin, R.; Peluso, S.; Nguyen, P.L.; Loda, M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010, 6, 551–562. [Google Scholar] [CrossRef] [Green Version]
- Landree, L.E.; Hanlon, A.L.; Strong, D.W.; Rumbaugh, G.; Miller, I.M.; Thupari, J.N.; Connolly, E.C.; Huganir, R.L.; Richardson, C.; Witters, L.A.; et al. C75, a fatty acid synthase inhibitor, modulates AMP-activated protein kinase to alter neuronal energy metabolism. J. Biol. Chem. 2004, 279, 3817–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronnett, G.V.; Kim, E.K.; Landree, L.E.; Tu, Y. Fatty acid metabolism as a target for obesity treatment. Physiol. Behav. 2005, 85, 25–35. [Google Scholar] [CrossRef]
- Gaunt, E.R.; Cheung, W.; Richards, J.E.; Lever, A.; Desselberger, U. Inhibition of rotavirus replication by downregulation of fatty acid synthesis. J. Gen. Virol. 2013, 94 Pt 6, 1310–1317. [Google Scholar] [CrossRef] [Green Version]
- Ohol, Y.M.; Wang, Z.; Kemble, G.; Duke, G. Direct inhibition of cellular fatty acid synthase impairs replication of respiratory syncytial virus and other respiratory viruses. PLoS ONE 2015, 10, e0144648. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Webster-Cyriaque, J.; Tomlinson, C.C.; Yohe, M.; Kenney, S. Fatty acid synthase expression is induced by the Epstein-Barr virus immediate-early protein BRLF1 and is required for lytic viral gene expression. J. Virol. 2004, 78, 4197–4206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rassmann, A.; Henke, A.; Jarasch, N.; Lottspeich, F.; Saluz, H.P.; Munder, T. The human fatty acid synthase: A new therapeutic target for coxsackievirus B3-induced diseases? Antivir. Res. 2007, 76, 150–158. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igal, R.A. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Biochim. Biophys. Acta 2016, 1861 Pt A, 1865–1880. [Google Scholar] [CrossRef]
- Uto, Y. Recent progress in the discovery and development of stearoyl CoA desaturase inhibitors. Chem. Phys. Lipids 2016, 197, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Lyn, R.K.; Singaravelu, R.; Kargman, S.; O’Hara, S.; Chan, H.; Oballa, R.; Huang, Z.; Jones, D.M.; Ridsdale, A.; Russell, R.S.; et al. Stearoyl-CoA desaturase inhibition blocks formation of hepatitis C virus-induced specialized membranes. Sci. Rep. 2014, 4, 4549. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.N.; Lim, Y.S.; Pham, L.V.; Shin, H.Y.; Kim, Y.S.; Hwang, S.B. Stearoyl coenzyme A desaturase 1 is associated with hepatitis C virus replication complex and regulates viral replication. J. Virol. 2014, 88, 12311–12325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nio, Y.; Hasegawa, H.; Okamura, H.; Miyayama, Y.; Akahori, Y.; Hijikata, M. Liver-specific mono-unsaturated fatty acid synthase-1 inhibitor for anti-hepatitis C treatment. Antivir. Res. 2016, 132, 262–267. [Google Scholar] [CrossRef]
- Hishiki, T.; Kato, F.; Nio, Y.; Watanabe, S.; Wen Tan, N.W.; Yamane, D.; Miyazaki, Y.; Lin, C.C.; Suzuki, R.; Tajima, S.; et al. Stearoyl-CoA desaturase-1 is required for flavivirus RNA replication. Antivir. Res. 2019, 165, 42–46. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting glutaminolysis: New perspectives to understand cancer development and novel strategies for potential target therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicker, C.A.; Hunt, B.G.; Krishnan, S.; Aziz, K.; Parajuli, S.; Palackdharry, S.; Elaban, W.R.; Wise-Draper, T.M.; Mills, G.B.; Waltz, S.E.; et al. Glutaminase inhibition with telaglenastat (CB-839) improves treatment response in combination with ionizing radiation in head and neck squamous cell carcinoma models. Cancer Lett. 2021, 502, 180–188. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef] [Green Version]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Ting Wu, T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef]
- Kim, K.; Kim, K.H.; Kim, H.H.; Cheong, J. Hepatitis B virus X protein induces lipogenic transcription factor SREBP1 and fatty acid synthase through the activation of nuclear receptor LXRalpha. Biochem. J. 2008, 416, 219–230. [Google Scholar] [CrossRef]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, E.A.; Chung, R.T. HCV and host lipids: An intimate connection. Semin. Liver Dis. 2013, 33, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Sidorkiewicz, M. Hepatitis C virus uses host lipids to its own advantage. Metabolites 2021, 11, 273. [Google Scholar] [CrossRef]
- Lavie, M.; Dubuisson, J. Interplay between hepatitis C virus and lipid metabolism during virus entry and assembly. Biochimie 2017, 141, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Blaising, J.; Pécheur, E.I. Lipids: A key for hepatitis C virus entry and a potential target for antiviral strategies. Biochimie 2013, 95, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ling, N.; Lei, Y.; Peng, M.; Hu, P.; Chen, M. Multifaceted interaction between Hepatitis B virus infection and lipid metabolism in hepatocytes: A potential target of antiviral therapy for chronic Hepatitis B. Front. Microbiol. 2021, 12, 636897. [Google Scholar] [CrossRef]
- Wang, C.C.; Tseng, T.C.; Kao, J.H. Hepatitis B virus infection and metabolic syndrome: Fact or fiction? J. Gastroenterol. Hepatol. 2015, 30, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Harwood, S.; Wise, L.M.; Purdy, J.G. Human cytomegalovirus pUL37x1 is important for remodeling of host lipid metabolism. J. Virol. 2019, 93, e00843-19. [Google Scholar] [CrossRef] [Green Version]
- Pirat, C.; Farce, A.; Lebègue, N.; Renault, N.; Furman, C.; Millet, R.; Yous, S.; Speca, S.; Berthelot, P.; Desreumaux, P.; et al. Targeting peroxisome proliferator-activated receptors (PPARs): Development of modulators. J. Med. Chem. 2012, 55, 4027–4061. [Google Scholar] [CrossRef]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and development of PPAR modulators in health and disease: An update of clinical evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammazzalorso, A.; Maccallini, C.; Amoia, P.; Amoroso, R. Multitarget PPARγ agonists as innovative modulators of the metabolic syndrome. Eur. J. Med. Chem. 2019, 173, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; Carrieri, A.; Verginelli, F.; Bruno, I.; Carbonara, G.; D’Angelo, A.; De Filippis, B.; Fantacuzzi, M.; Florio, R.; Fracchiolla, G.; et al. Synthesis, in vitro evaluation, and molecular modeling investigation of benzenesulfonimide peroxisome proliferator-activated receptors α antagonists. Eur. J. Med. Chem. 2016, 114, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, E.; d’Angelo, M.; Ammazzalorso, A.; Gravina, G.L.; Laezza, C.; Antonosante, A.; Panella, G.; Cinque, B.; Cristiano, L.; Dhez, A.C.; et al. PPARα antagonist AA452 triggers metabolic reprogramming and increases sensitivity to radiation therapy in human glioblastoma primary cells. J. Cell Physiol. 2017, 232, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; Bruno, I.; Florio, R.; De Lellis, L.; Laghezza, A.; Cerchia, C.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; et al. Sulfonimide and amide derivatives as novel PPARα antagonists: Synthesis, antiproliferative activity, and docking studies. ACS Med. Chem. Lett. 2020, 11, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Ammazzalorso, A.; De Lellis, L.; Florio, R.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Perconti, S.; Verginelli, F.; et al. Cytotoxic effect of a family of peroxisome proliferator-activated receptor antagonists in colorectal and pancreatic cancer cell lines. Chem. Biol. Drug Des. 2017, 90, 1029–1035. [Google Scholar] [CrossRef]
- De Lellis, L.; Cimini, A.; Veschi, S.; Benedetti, E.; Amoroso, R.; Cama, A.; Ammazzalorso, A. The anticancer potential of Peroxisome Proliferator-Activated Receptor antagonists. ChemMedChem 2018, 13, 209–219. [Google Scholar] [CrossRef]
- Yan, B.; Chu, H.; Yang, D.; Sze, K.H.; Lai, P.M.; Yuan, S.; Shuai, H.; Wang, Y.; Kao, R.Y.; Chan, J.F.; et al. Characterization of the lipidomic profile of human coronavirus-infected cells: Implications for lipid metabolism remodeling upon coronavirus replication. Viruses 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Li, H.; Hu, S.; Zhou, Y. ACE2 correlated with immune infiltration serves as a prognostic biomarker in endometrial carcinoma and renal papillary cell carcinoma: Implication for COVID-19. Aging 2020, 12, 6518–6535. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, L.; Li, M.; Wang, X. The SARS-CoV-2 host cell receptor ACE2 correlates positively with immunotherapy response and is a potential protective factor for cancer progression. Comput. Struct. Biotechnol. J. 2020, 18, 2438–2444. [Google Scholar] [CrossRef]
- Heffernan, K.S.; Ranadive, S.M.; Jae, S.Y. Exercise as medicine for COVID-19: On PPAR with emerging pharmacotherapy. Med. Hypotheses 2020, 143, 110197. [Google Scholar] [CrossRef]
- Ehrlich, A.; Uhl, S.; Ioannidis, K.; Hofree, M.; ten Oever, B.; Nahmias, Y. The SARS-CoV-2 Transcriptional Metabolic Signature in Lung Epithelium. SSRN Electron J. 2020. [Google Scholar] [CrossRef]
- Rodon, J.; Muñoz-Basagoiti, J.; Perez-Zsolt, D.; Noguera-Julian, M.; Paredes, R.; Mateu, L.; Quiñones, C.; Perez, C.; Erkizia, I.; Blanco, I.; et al. Identification of plitidepsin as potent inhibitor of SARS-CoV-2-induced cytopathic effect after a drug repurposing screen. Front. Pharmacol. 2021, 12, 646676. [Google Scholar] [CrossRef]
- Del Re, A.; Corpetti, C.; Pesce, M.; Seguella, L.; Steardo, L.; Palenca, I.; Rurgo, S.; De Conno, B.; Sarnelli, G.; Esposito, G. Ultramicronized Palmitoylethanolamide inhibits NLRP3 inflammasome expression and pro-inflammatory response activated by SARS-CoV-2 Spike protein in cultured murine alveolar macrophages. Metabolites 2021, 11, 592. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Interplay of opposing effects of the WNT/β-Catenin pathway and PPARγ and implications for SARS-CoV2 treatment. Front. Immunol. 2021, 12, 666693. [Google Scholar] [CrossRef]
- Ciavarella, C.; Motta, I.; Valente, S.; Pasquinelli, G. Pharmacological (or synthetic) and nutritional agonists of PPAR-γ as candidates for cytokine storm modulation in COVID-19 disease. Molecules 2020, 25, 2076. [Google Scholar] [CrossRef] [PubMed]
- Jagat, J.M.; Kalyan, K.G.; Subir, R. Use of pioglitazone in people with type 2 diabetes mellitus with coronavirus disease 2019 (COVID-19): Boon or bane? Diabetes Metab. Syndr. 2020, 14, 829–831. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Carboni, E.; Carta, A.R.; Carboni, E. Can pioglitazone be potentially useful therapeutically in treating patients with COVID-19? Med. Hypotheses 2020, 140, 109776. [Google Scholar] [CrossRef]
- Francisqueti-Ferron, F.V.; Garcia, J.L.; Ferron, A.J.T.; Nakandakare-Maia, E.T.; Gregolin, C.S.; Silva, J.P.D.C.; Dos Santos, K.C.; Lo, Â.T.C.; Siqueira, J.S.; de Mattei, L.; et al. Gamma-oryzanol as a potential modulator of oxidative stress and inflammation via PPAR-γ in adipose tissue: A hypothetical therapeutic for cytokine storm in COVID-19? Mol. Cell Endocrinol. 2021, 520, 111095. [Google Scholar] [CrossRef]
- Talukdar, J.; Bhadra, B.; Dattaroy, T.; Nagle, V.; Dasgupta, S. Potential of natural astaxanthin in alleviating the risk of cytokine storm in COVID-19. Biomed. Pharmacother. 2020, 132, 110886. [Google Scholar] [CrossRef] [PubMed]
- AbdelMassih, A.F.; Menshawey, R.; Ismail, J.H.; Husseiny, R.J.; Husseiny, Y.M.; Yacoub, S.; Kamel, A.; Hozaien, R.; Yacoub, E.; Menshawey, E.; et al. PPAR agonists as effective adjuvants for COVID-19 vaccines, by modifying immunogenetics: A review of literature. J. Genet. Eng. Biotechnol. 2021, 19, 82. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yang, J.; Lee, S.L.; Kulp, S.K.; Chen, C.S. PPARgamma-independent antitumor effects of thiazolidinediones. Cancer Lett. 2009, 276, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, M.; Yagami, T.; Fujio, M.; Tohji, C.; Takase, K.; Yamamoto, Y.; Sawada, K.; Yamamori, M.; Okamura, N. Cytotoxicity of troglitazone through PPARγ-independent pathway and p38 MAPK pathway in renal cell carcinoma. Cancer Lett. 2011, 312, 219–227. [Google Scholar] [CrossRef]
- Yamasaki, D.; Kawabe, N.; Nakamura, H.; Tachibana, K.; Ishimoto, K.; Tanaka, T.; Aburatani, H.; Sakai, J.; Hamakubo, T.; Kodama, T.; et al. Fenofibrate suppresses growth of the human hepatocellular carcinoma cell via PPARα-independent mechanisms. Eur. J. Cell Biol. 2011, 90, 657–664. [Google Scholar] [CrossRef]
- Davidson, M.A.; Mattison, D.R.; Azoulay, L.; Krewski, D. Thiazolidinedione drugs in the treatment of type 2 diabetes mellitus: Past, present and future. Crit. Rev. Toxicol. 2018, 48, 52–108. [Google Scholar] [CrossRef]
- Dharancy, S.; Lemoine, M.; Mathurin, P.; Serfaty, L.; Dubuquoy, L. Peroxisome proliferator-activated receptors in HCV-related infection. PPAR Res. 2009, 2009, 357204. [Google Scholar] [CrossRef] [Green Version]
- Dharancy, S.; Malapel, M.; Perlemuter, G.; Roskams, T.; Cheng, Y.; Dubuquoy, L.; Podevin, P.; Conti, F.; Canva, V.; Philippe, D.; et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology 2005, 128, 334–342. [Google Scholar] [CrossRef] [Green Version]
- de Gottardi, A.; Pazienza, V.; Pugnale, P.; Bruttin, F.; Rubbia-Brandt, L.; Juge-Aubry, C.E.; Meier, C.A.; Hadengue, A.; Negro, F. Peroxisome proliferator-activated receptor-alpha and -gamma mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment. Pharmacol. Ther. 2006, 23, 107–114. [Google Scholar] [CrossRef]
- Kim, K.; Kim, K.H.; Ha, E.; Park, J.Y.; Sakamoto, N.; Cheong, J. Hepatitis C virus NS5A protein increases hepatic lipid accumulation via induction of activation and expression of PPARgamma. FEBS Lett. 2009, 583, 2720–2726. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, T.; Niimi, A.; Maeda, A.; Shigemoto, M.; Yamashita, K. Study of effectiveness of bezafibrate in the treatment of chronic hepatitis C. Am. J. Gastroenterol. 2001, 96, 1659–1660. [Google Scholar] [CrossRef]
- Fujita, N.; Kaito, M.; Tanaka, H.; Horiike, S.; Adachi, Y. Reduction of serum HCV RNA titer by bezafibrate therapy in patients with chronic hepatitis C. Am. J. Gastroenterol. 2004, 99, 2280. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Kaito, M.; Kai, M.; Sugimoto, R.; Tanaka, H.; Horiike, S.; Konishi, M.; Iwasa, M.; Watanabe, S.; Adachi, Y. Effects of bezafibrate in patients with chronic hepatitis C virus infection: Combination with interferon and ribavirin. J. Viral Hepat. 2006, 13, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Chojkier, M.; Elkhayat, H.; Sabry, D.; Donohue, M.; Buck, M. Pioglitazone decreases hepatitis C viral load in overweight, treatment naïve, genotype 4 infected-patients: A pilot study. PLoS ONE 2012, 7, e31516. [Google Scholar] [CrossRef] [Green Version]
- Goldwasser, J.; Cohen, P.Y.; Lin, W.; Kitsberg, D.; Balaguer, P.; Polyak, S.J.; Chung, R.T.; Yarmush, M.L.; Nahmias, Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J. Hepatol. 2011, 55, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.C.; Bai, C.H.; Su, H.C.; Tsai, P.J.; Pu, C.Y.; Liao, C.S.; Lin, Y.M.; Lai, H.W.; Chong, L.W.; Tsai, Y.S.; et al. Fluoxetine a novel anti-hepatitis C virus agent via ROS-, JNK-, and PPARβ/γ-dependent pathways. Antivir. Res. 2014, 110, 158–167. [Google Scholar] [CrossRef]
- Lin, Y.M.; Sun, H.Y.; Chiu, W.T.; Su, H.C.; Chien, Y.C.; Chong, L.W.; Chang, H.C.; Bai, C.H.; Young, K.C.; Tsao, C.W. Calcitriol inhibits HCV infection via blockade of activation of PPAR and interference with endoplasmic reticulum-associated degradation. Viruses 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Rakic, B.; Sagan, S.M.; Noestheden, M.; Bélanger, S.; Nan, X.; Evans, C.L.; Xie, X.S.; Pezacki, J.P. Peroxisome proliferator-activated receptor alpha antagonism inhibits hepatitis C virus replication. Chem. Biol. 2006, 13, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ban, S.; Ueda, Y.; Ohashi, M.; Matsuno, K.; Ikeda, M.; Kato, N.; Miyachi, H. Peroxisome proliferator-activated receptor delta antagonists inhibit hepatitis C virus RNA replication. Bioorg. Med. Chem. Lett. 2013, 23, 4774–4778. [Google Scholar] [CrossRef]
- Rauwel, B.; Mariamé, B.; Martin, H.; Nielsen, R.; Allart, S.; Pipy, B.; Mandrup, S.; Devignes, M.D.; Evain-Brion, D.; Fournier, T.; et al. Activation of peroxisome proliferator-activated receptor gamma by human cytomegalovirus for de novo replication impairs migration and invasiveness of cytotrophoblasts from early placentas. J. Virol. 2010, 84, 2946–2954. [Google Scholar] [CrossRef] [Green Version]
- Leghmar, K.; Cenac, N.; Rolland, M.; Martin, H.; Rauwel, B.; Bertrand-Michel, J.; Le Faouder, P.; Bénard, M.; Casper, C.; Davrinche, C.; et al. Cytomegalovirus infection triggers the secretion of the PPARγ agonists 15-hydroxyeicosatetraenoic acid (15-HETE) and 13-hydroxyoctadecadienoic acid (13-HODE) in human cytotrophoblasts and placental cultures. PLoS ONE 2015, 10, e0132627. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Li, X.; Sellier, Y.; Martin, H.; Perez-Berezo, T.; Rauwel, B.; Benchoua, A.; Bessières, B.; Aziza, J.; Cenac, N.; et al. PPARγ is activated during congenital cytomegalovirus infection and inhibits neuronogenesis from human neural stem cells. PLoS Pathog. 2016, 12, e1005547. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | PPAR Activity | Concentration | Cell | Outcome | Ref. |
|---|---|---|---|---|---|
| Fenofibrate (Tricor®) | PPARα agonist | 20 µM | Human bronchial epithelial cells | Block viral replication, reverted effects on phospholipid accumulation and glycolysis | [65] |
| Fenofibrate | PPARα agonist | 20 µM | Vero E6 cells | Block viral entry | [66] |
| Palmitoylethanolamide | PPARα agonist | 10−9–10−7 M | murine alveolar macrophages | Inhibition of NF-κB transcription and NLRP-3 inflammasome signaling, with a significant antinflammatory effect | [67] |
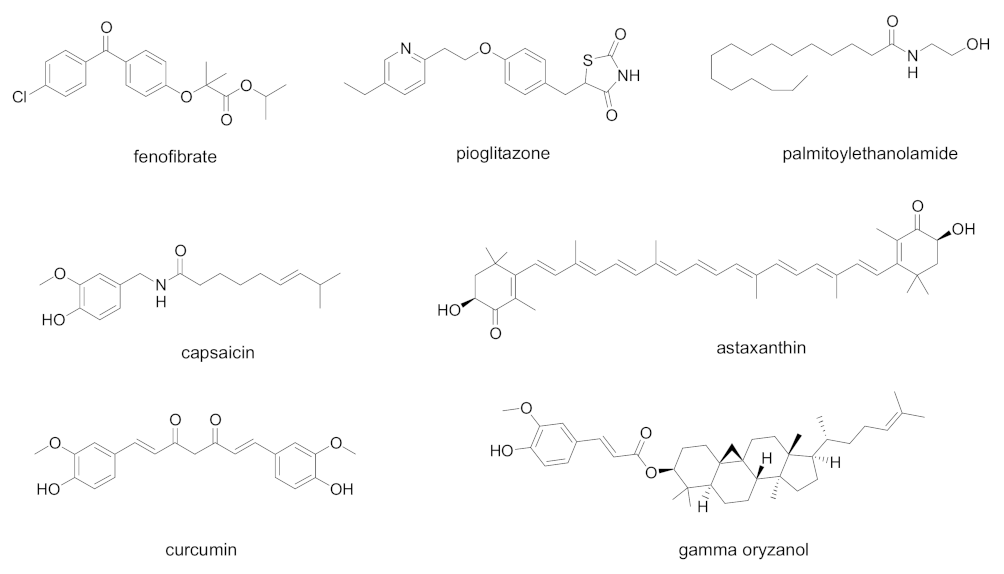
| Pioglitazone | PPARγ agonist | - | - | Proposed as 3CL-Pro inhibitor, it could downregulate SARS-CoV-2 RNA synthesis and replication Proposed as an additive in COVID-19 patients with diabetes, hypertension, and cardiovascular comorbidities for its antinflammatory properties | [71,72] |
| Curcumin, capsaicin, docosahexanoic acid, eicosapentaenoic acid | Natural PPARγ agonists | - | - | Proposed for use in COVID-19, due to their ability to prevent cytokine overproduction and inflammatory cascade | [69] |
| Gamma-oryzanol | PPARγ modulator | - | - | Proposed for use in COVID-19, due to its anti- inflammatory and antioxidant properties | [73] |
| Astaxanthin | Multiple action on PPARs: PPARα agonist, PPARδ antagonist, PPARγ agonist or antagonist | - | - | Proposed for use in COVID-19, due to the ability to reduce the oxidative stress induced by ROS, the immune response, and the production of pro-inflammatory cytokines | [74] |
| Cpd | PPAR Activity | Concentration | Model | Outcome | Ref. |
|---|---|---|---|---|---|
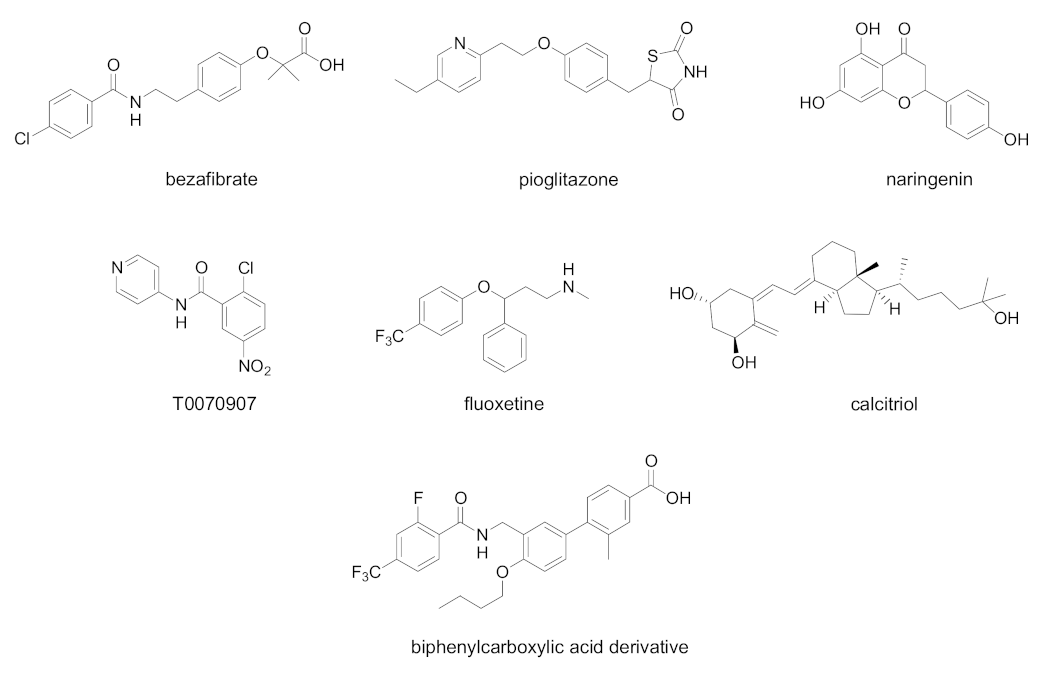
| Bezafibrate | PPAR pan agonist | 400 mg/day for 8 weeks | Chronic hepatitis C patients complicated with hyperlipidemia | Decreased serum HCV RNA | [86] |
| Pioglitazone | PPARγ agonist | 30 mg/day for 14 days | Overweight Genotype 4 HCV patients | Decreased serum HCV RNA at day 14 | [87] |
| Naringenin | PPARα agonist | 200 µM | HCV-infected Huh7.5.1 | Inhibition of ApoB-100 and HCV RNA secretion | [88] |
| Fluoxetine | PPARγ/δ modulator | 0.1–10 µM for 6 days | HCV-infected Huh7.5 cells | Decrease in virus protein levels of core, NS3, and NS5A. Reduction in ROS generation and lipid accumulation | [89] |
| Calcitriol | PPARα/γ/δ modulator | 0.1–1000 nM | HCV-infected Huh7.5 cells | Decrease in viral infection, nitrative stress, and lipid accumulation | [90] |
| T0070907 | PPARα/γ antagonist | IC50 19.1 µM | Huh-7 cells expressing an HCV subgenomic replicon | Inhibition of HCV replication | [91] |
| Biphenylcarboxylic acids | PPARδ antagonists | 2.5–10.0 µM, most potent compound EC50 0.22 µM | OR6 HCV replication system | Dose-dependent inhibition of HCV RNA replication. Synergistic antiviral effect when tested in combination with Peg-IFNα or Peg-IFNα and ribavirin | [92] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fantacuzzi, M.; Amoroso, R.; Ammazzalorso, A. PPAR Ligands Induce Antiviral Effects Targeting Perturbed Lipid Metabolism during SARS-CoV-2, HCV, and HCMV Infection. Biology 2022, 11, 114. https://doi.org/10.3390/biology11010114
Fantacuzzi M, Amoroso R, Ammazzalorso A. PPAR Ligands Induce Antiviral Effects Targeting Perturbed Lipid Metabolism during SARS-CoV-2, HCV, and HCMV Infection. Biology. 2022; 11(1):114. https://doi.org/10.3390/biology11010114
Chicago/Turabian StyleFantacuzzi, Marialuigia, Rosa Amoroso, and Alessandra Ammazzalorso. 2022. "PPAR Ligands Induce Antiviral Effects Targeting Perturbed Lipid Metabolism during SARS-CoV-2, HCV, and HCMV Infection" Biology 11, no. 1: 114. https://doi.org/10.3390/biology11010114