
PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH)

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
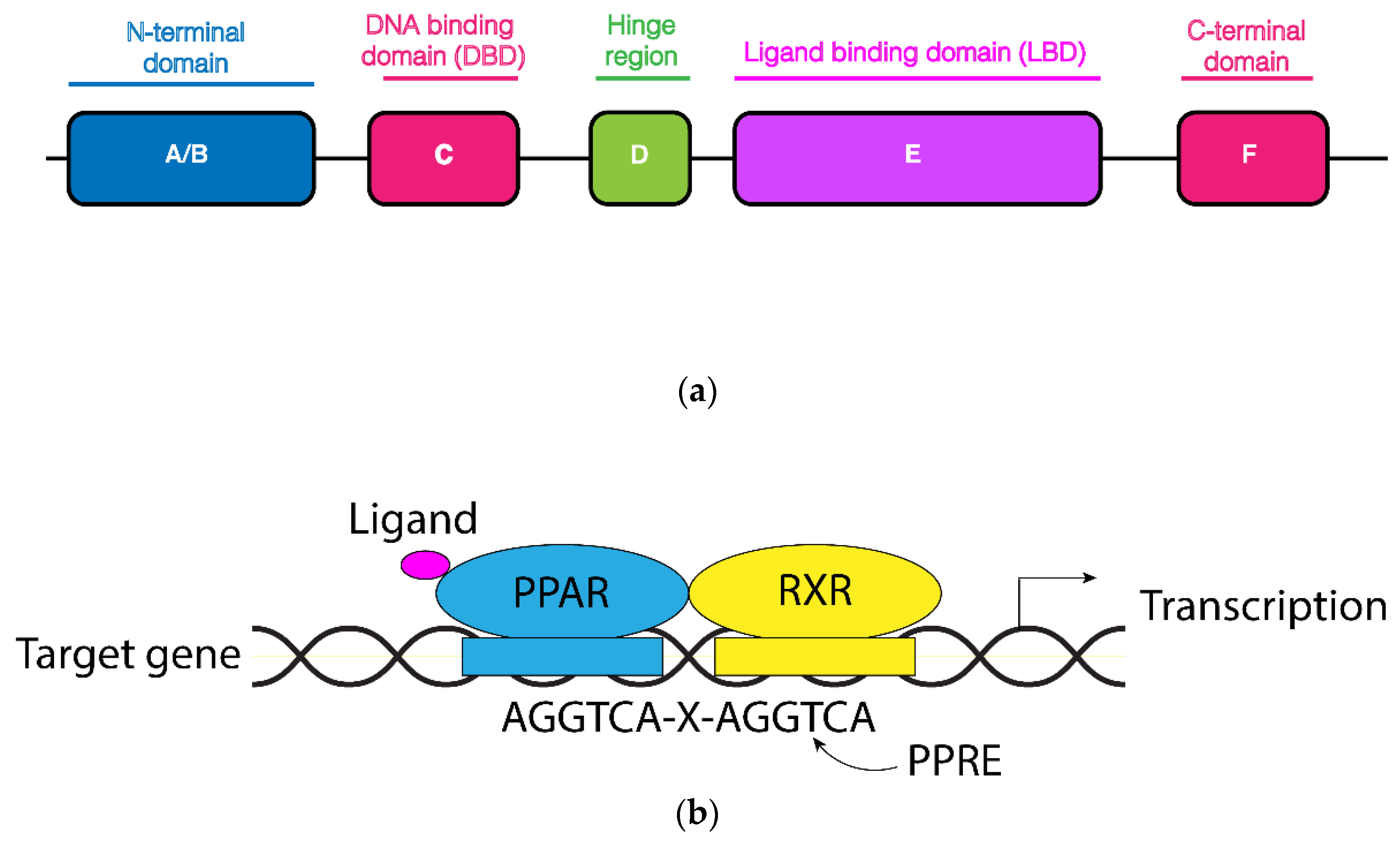
2. Main Structural and Functional Features
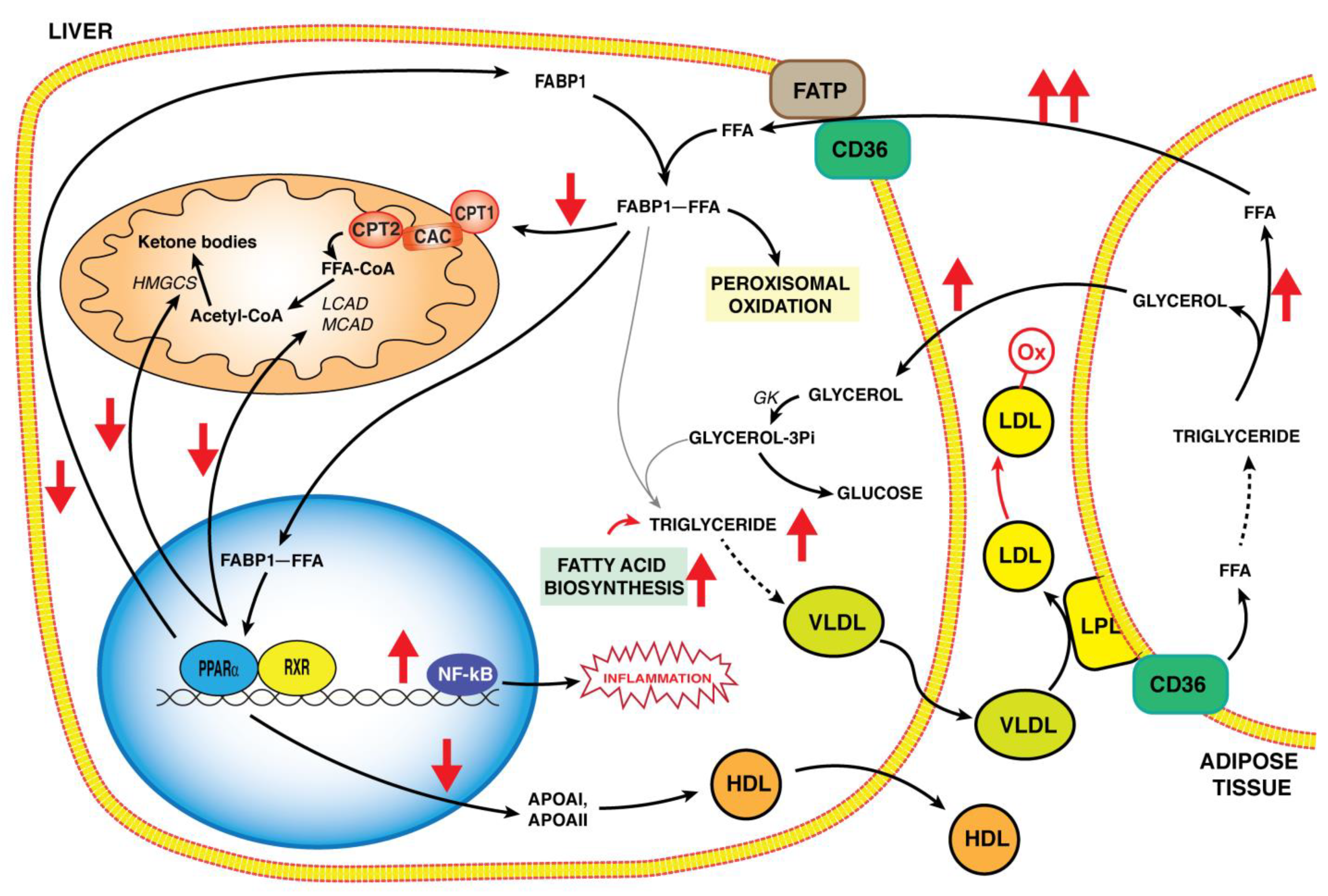
3. PPARα and Liver Metabolism
4. PPARα as a Modulator of Inflammatory Pathways
5. PPARα Expression in NASH Liver
6. Role of PPARα in the Pathogenesis of NASH
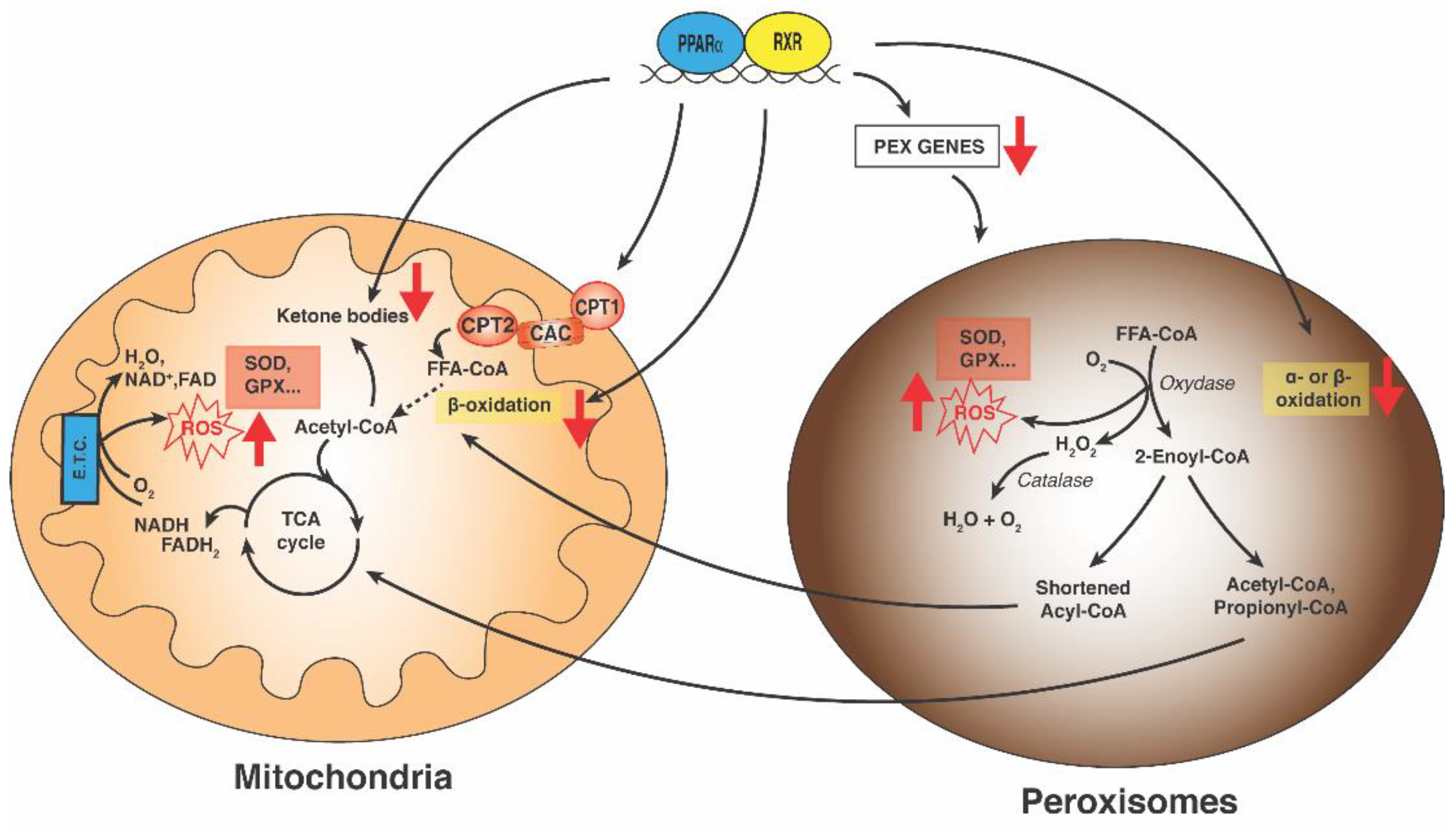
7. PPARα and Peroxisomes in NASH
8. PPARα and Mitochondria in NASH
9. Targeting PPARα as Therapy for NASH
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Hui, J.M.; Marchesini, G.; Bugianesi, E.; George, J.; Farrell, G.C.; Enders, F.; Saksena, S.; Burt, A.D.; Bida, J.P.; et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007, 45, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Barger, P.M.; Browning, A.C.; Garner, A.N.; Kelly, D.P. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor alpha: A potential role in the cardiac metabolic stress response. J. Biol. Chem. 2001, 276, 44495–44501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gearing, K.L.; Crickmore, A.; Gustafsson, J.A. Structure of the mouse peroxisome proliferator activated receptor alpha gene. Biochem. Biophys. Res. Commun. 1994, 199, 255–263. [Google Scholar] [CrossRef]
- Staels, B.; Maes, M.; Zambon, A. Fibrates and future PPARalpha agonists in the treatment of cardiovascular disease. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 542–553. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauca, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Mandard, S.; Voshol, P.J.; Escher, P.; Tan, N.S.; Havekes, L.M.; Koenig, W.; Marz, W.; Tafuri, S.; Wahli, W.; et al. PPARalpha governs glycerol metabolism. J. Clin. Investig. 2004, 114, 94–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.S.; Kim, J. Peroxisome Proliferator-Activated Receptors and the Heart: Lessons from the Past and Future Directions. PPAR Res. 2015, 2015, 271983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors (PPARs): Nuclear receptors at the crossroads between lipid metabolism and inflammation. Inflamm. Res. 2000, 49, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Mandard, S.; Escher, P.; Gonzalez, F.J.; Tafuri, S.; Desvergne, B.; Wahli, W. The peroxisome proliferator-activated receptor alpha regulates amino acid metabolism. FASEB J. 2001, 15, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Montagner, A.; Polizzi, A.; Fouche, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Regnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Suiter, C.; Singha, S.K.; Khalili, R.; Shariat-Madar, Z. Free Fatty Acids: Circulating Contributors of Metabolic Syndrome. Cardiovasc. Hematol. Agents Med. Chem. 2018, 16, 20–34. [Google Scholar] [CrossRef]
- Marra, F.; Lotersztajn, S. Pathophysiology of NASH: Perspectives for a targeted treatment. Curr. Pharm. Des. 2013, 19, 5250–5269. [Google Scholar] [CrossRef] [Green Version]
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonekamp, N.A.; Volkl, A.; Fahimi, H.D.; Schrader, M. Reactive oxygen species and peroxisomes: Struggling for balance. Biofactors 2009, 35, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef] [PubMed]
- Sandalio, L.M.; Rodriguez-Serrano, M.; Romero-Puertas, M.C.; del Rio, L.A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell. Biochem. 2013, 69, 231–255. [Google Scholar] [PubMed]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef]
- Chawla, A.; Repa, J.J.; Evans, R.M.; Mangelsdorf, D.J. Nuclear receptors and lipid physiology: Opening the X-files. Science 2001, 294, 1866–1870. [Google Scholar] [CrossRef] [Green Version]
- Gronemeyer, H.; Laudet, V. Transcription factors 3: Nuclear receptors. Protein Profile 1995, 2, 1173–1308. [Google Scholar]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Santos, R.D.; Aguilar-Salinas, C.; Aikawa, M.; Al Rasadi, K.; Amarenco, P.; Barter, P.J.; Ceska, R.; Corsini, A.; Despres, J.P.; et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha) paradigm: Conceptual framework and therapeutic potential: A consensus statement from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) Foundation. Cardiovasc. Diabetol. 2019, 18, 71. [Google Scholar]
- Georgiadi, A.; Kersten, S. Mechanisms of gene regulation by fatty acids. Adv. Nutr. 2012, 3, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, V.; Metcalf, J. Metabolic functions of the liver. Anaesth. Intensive Care Med. 2009, 10, 334–335. [Google Scholar] [CrossRef]
- Infantino, V.; Dituri, F.; Convertini, P.; Santarsiero, A.; Palmieri, F.; Todisco, S.; Mancarella, S.; Giannelli, G.; Iacobazzi, V. Epigenetic upregulation and functional role of the mitochondrial aspartate/glutamate carrier isoform 1 in hepatocellular carcinoma. Biochim. Biophys. Acta Mol. Basis Dis 2019, 1865, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Infantino, V.; Santarsiero, A.; Convertini, P.; Todisco, S.; Iacobazzi, V. Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 5703. [Google Scholar] [CrossRef]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 1994, 79, 427–436. [Google Scholar] [CrossRef]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Atshaves, B.P.; Martin, G.G.; Hostetler, H.A.; McIntosh, A.L.; Kier, A.B.; Schroeder, F. Liver fatty acid-binding protein and obesity. J. Nutr. Biochem. 2010, 21, 1015–1032. [Google Scholar] [CrossRef] [Green Version]
- Ong, K.T.; Mashek, M.T.; Davidson, N.O.; Mashek, D.G. Hepatic ATGL mediates PPAR-alpha signaling and fatty acid channeling through an L-FABP independent mechanism. J. Lipid Res. 2014, 55, 808–815. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Peters, J.M.; Iritani, N.; Nakajima, T.; Furihata, K.; Hashimoto, T.; Gonzalez, F.J. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 1998, 273, 5678–5684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulick, T.; Cresci, S.; Caira, T.; Moore, D.D.; Kelly, D.P. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc. Natl. Acad. Sci. USA 1994, 91, 11012–11016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougarne, N.; Weyers, B.; Desmet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular Actions of PPARalpha in Lipid Metabolism and Inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARalpha. Int. J. Mol. Sci. 2016, 17. [Google Scholar]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Schoonjans, K.; Peinado-Onsurbe, J.; Lefebvre, A.M.; Heyman, R.A.; Briggs, M.; Deeb, S.; Staels, B.; Auwerx, J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996, 15, 5336–5348. [Google Scholar] [CrossRef]
- Ferre, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 83–92. [Google Scholar] [CrossRef]
- Infantino, V.; Iacobazzi, V.; De Santis, F.; Mastrapasqua, M.; Palmieri, F. Transcription of the mitochondrial citrate carrier gene: Role of SREBP-1, upregulation by insulin and downregulation by PUFA. Biochem. Biophys. Res. Commun. 2007, 356, 249–254. [Google Scholar] [CrossRef]
- Fernandez-Alvarez, A.; Alvarez, M.S.; Gonzalez, R.; Cucarella, C.; Muntane, J.; Casado, M. Human SREBP1c expression in liver is directly regulated by peroxisome proliferator-activated receptor alpha (PPARalpha). J. Biol. Chem. 2011, 286, 21466–21477. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.D.; Knight, B.L.; Wiggins, D.; Humphreys, S.M.; Gibbons, G.F. Disturbances in the normal regulation of SREBP-sensitive genes in PPAR alpha-deficient mice. J. Lipid Res. 2001, 42, 328–337. [Google Scholar] [CrossRef]
- Oosterveer, M.H.; Grefhorst, A.; van Dijk, T.H.; Havinga, R.; Staels, B.; Kuipers, F.; Groen, A.K.; Reijngoud, D.J. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J. Biol Chem. 2009, 284, 34036–34044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, A. Control of macrophage activation and function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 2010, 10, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Ogawa, S. Combinatorial roles of nuclear receptors in inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Kostadinova, R.; Wahli, W.; Michalik, L. PPARs in diseases: Control mechanisms of inflammation. Curr. Med. Chem. 2005, 12, 2995–3009. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [Green Version]
- Diamant, G.; Dikstein, R. Transcriptional control by NF-kappaB: Elongation in focus. Biochim. Biophys. Acta 2013, 1829, 937–945. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Mattioli, I.; Buss, H.; Kracht, M. NF-kappaB: A multifaceted transcription factor regulated at several levels. Chembiochem 2004, 5, 1348–1358. [Google Scholar] [CrossRef]
- Mochizuki, K.; Suzuki, T.; Goda, T. PPAR alpha and PPAR delta transactivity and p300 binding activity induced by arachidonic acid in colorectal cancer cell line Caco-2. J. Nutr. Sci. Vitaminol. 2008, 54, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Santarsiero, A.; Convertini, P.; Todisco, S.; Pierri, C.L.; De Grassi, A.; Williams, N.C.; Iacobazzi, D.; De Stefano, G.; O’Neill, L.A.J.; Infantino, V. ACLY Nuclear Translocation in Human Macrophages Drives Proinflammatory Gene Expression by NF-kappaB Acetylation. Cells 2021, 10, 2962. [Google Scholar] [CrossRef]
- Wang, W.; Lin, Q.; Lin, R.; Zhang, J.; Ren, F.; Zhang, J.; Ji, M.; Li, Y. PPARalpha agonist fenofibrate attenuates TNF-alpha-induced CD40 expression in 3T3-L1 adipocytes via the SIRT1-dependent signaling pathway. Exp. Cell Res. 2013, 319, 1523–1533. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Bai, L.; Qiao, H.; Lu, Y.; Yang, L.; Zhang, J.; Lin, R.; Ren, F.; Zhang, J.; Ji, M. The protective effect of fenofibrate against TNF-alpha-induced CD40 expression through SIRT1-mediated deacetylation of NF-kappaB in endothelial cells. Inflammation 2014, 37, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; De Bosscher, K.; Vanden Berghe, W.; Fruchart, J.C.; Haegeman, G.; Staels, B. DNA binding-independent induction of IkappaBalpha gene transcription by PPARalpha. Mol. Endocrinol. 2002, 16, 1029–1039. [Google Scholar]
- Delerive, P.; Gervois, P.; Fruchart, J.C.; Staels, B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J. Biol. Chem. 2000, 275, 36703–36707. [Google Scholar] [PubMed] [Green Version]
- Grau, R.; Punzon, C.; Fresno, M.; Iniguez, M.A. Peroxisome-proliferator-activated receptor alpha agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem. J. 2006, 395, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Joe, E.H.; Jou, I. PPAR-alpha activators suppress STAT1 inflammatory signaling in lipopolysaccharide-activated rat glia. Neuroreport 2005, 16, 829–833. [Google Scholar]
- Shipley, J.M.; Waxman, D.J. Down-regulation of STAT5b transcriptional activity by ligand-activated peroxisome proliferator-activated receptor (PPAR) alpha and PPARgamma. Mol. Pharmacol. 2003, 64, 355–364. [Google Scholar] [CrossRef]
- Shipley, J.M.; Waxman, D.J. Simultaneous, bidirectional inhibitory crosstalk between PPAR and STAT5b. Toxicol. Appl. Pharmacol. 2004, 199, 275–284. [Google Scholar] [CrossRef]
- Poynter, M.E.; Daynes, R.A. Peroxisome proliferator-activated receptor alpha activation modulates cellular redox status, represses nuclear factor-kappaB signaling, and reduces inflammatory cytokine production in aging. J. Biol. Chem. 1998, 273, 32833–32841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Woerly, G.; Honda, K.; Loyens, M.; Papin, J.P.; Auwerx, J.; Staels, B.; Capron, M.; Dombrowicz, D. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J. Exp. Med. 2003, 198, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Ouyang, W. The function role of GATA-3 in Th1 and Th2 differentiation. Immunol. Res. 2003, 28, 25–37. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Ward, K.W. WY-14 643, a selective PPAR{alpha} agonist, induces proinflammatory and proangiogenic responses in human ocular cells. Int. J. Toxicol. 2010, 29, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shi, W.; Tontonoz, P.; Wang, S.; Subbanagounder, G.; Hedrick, C.C.; Hama, S.; Borromeo, C.; Evans, R.M.; Berliner, J.A.; et al. Role for peroxisome proliferator-activated receptor alpha in oxidized phospholipid-induced synthesis of monocyte chemotactic protein-1 and interleukin-8 by endothelial cells. Circ. Res. 2000, 87, 516–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.R.; Clarke, S.; Rodgers, K.; Thornhill, B.; Peters, J.M.; Gonzalez, F.J.; Gimble, J.M. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect. Immun. 1999, 67, 3488–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colville-Nash, P.R.; Qureshi, S.S.; Willis, D.; Willoughby, D.A. Inhibition of inducible nitric oxide synthase by peroxisome proliferator-activated receptor agonists: Correlation with induction of heme oxygenase 1. J. Immunol. 1998, 161, 978–984. [Google Scholar]
- Rakhshandehroo, M.; Hooiveld, G.; Muller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Bayha, C.; Klein, K.; Muller, S.; Weiss, T.S.; Schwab, M.; Zanger, U.M. The truncated splice variant of peroxisome proliferator-activated receptor alpha, PPARalpha-tr, autonomously regulates proliferative and pro-inflammatory genes. BMC Cancer 2015, 15, 488. [Google Scholar] [CrossRef] [Green Version]
- Westerbacka, J.; Kolak, M.; Kiviluoto, T.; Arkkila, P.; Siren, J.; Hamsten, A.; Fisher, R.M.; Yki-Jarvinen, H. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007, 56, 2759–2765. [Google Scholar] [CrossRef] [Green Version]
- Francque, S.; Verrijken, A.; Caron, S.; Prawitt, J.; Paumelle, R.; Derudas, B.; Lefebvre, P.; Taskinen, M.R.; Van Hul, W.; Mertens, I.; et al. PPARalpha gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J. Hepatol. 2015, 63, 164–173. [Google Scholar] [CrossRef]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, J.E.; Ko, M.S.; Woo, S.H.; Kim, B.J.; Kim, H.S.; Park, H.S.; Park, I.S.; Koh, E.H.; Lee, K.U. Statins Increase Mitochondrial and Peroxisomal Fatty Acid Oxidation in the Liver and Prevent Non-Alcoholic Steatohepatitis in Mice. Diabetes Metab. J. 2016, 40, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.S.; Ng, D.L.; Kor, S.B.; Wong, H.K.; Tengku-Muhammad, T.S.; Choo, Q.C.; Chew, C.H. Tumour necrosis factor alpha down-regulates the expression of peroxisome proliferator activated receptor alpha (PPARalpha) in human hepatocarcinoma HepG2 cells by activation of NF-kappaB pathway. Cytokine 2013, 61, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.H.; Chew, G.S.; Najimudin, N.; Tengku-Muhammad, T.S. Interleukin-6 inhibits human peroxisome proliferator activated receptor alpha gene expression via CCAAT/enhancer-binding proteins in hepatocytes. Int. J. Biochem. Cell Biol. 2007, 39, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Reddy, J.K.; Rao, M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Sohn, I.; Ahn, J.I.; Lee, K.H.; Lee, Y.S.; Lee, Y.S. Hepatic gene expression profiles in a long-term high-fat diet-induced obesity mouse model. Gene 2004, 340, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Reddy, J.K.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, C.; Soardo, G.; Esposito, W.; Fallo, F.; Basan, L.; Donnini, D.; Federspil, G.; Sechi, L.A.; Vettor, R. Plasma adiponectin is decreased in nonalcoholic fatty liver disease. Eur. J. Endocrinol. 2005, 152, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.M.; Sun, R.Q.; Zeng, X.Y.; Choong, Z.H.; Wang, H.; Watt, M.J.; Ye, J.M. Activation of PPARalpha ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress. Diabetes 2013, 62, 2095–2105. [Google Scholar] [CrossRef] [Green Version]
- Cook, W.S.; Jain, S.; Jia, Y.; Cao, W.Q.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Peroxisome proliferator-activated receptor alpha-responsive genes induced in the newborn but not prenatal liver of peroxisomal fatty acyl-CoA oxidase null mice. Exp. Cell Res. 2001, 268, 70–76. [Google Scholar] [CrossRef]
- Schrader, M.; Fahimi, H.D. Peroxisomes and oxidative stress. Biochim. Biophys. Acta 2006, 1763, 1755–1766. [Google Scholar] [CrossRef] [Green Version]
- Kakimoto, P.A.; Tamaki, F.K.; Cardoso, A.R.; Marana, S.R.; Kowaltowski, A.J. H2O2 release from the very long chain acyl-CoA dehydrogenase. Redox Biol. 2015, 4, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Piao, L.; Choi, J.; Kwon, G.; Ha, H. Endogenous catalase delays high-fat diet-induced liver injury in mice. Korean J. Physiol. Pharmacol. 2017, 21, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Waterham, H.R.; Ebberink, M.S. Genetics and molecular basis of human peroxisome biogenesis disorders. Biochim. Biophys. Acta 2012, 1822, 1430–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baes, M.; Van Veldhoven, P.P. Hepatic dysfunction in peroxisomal disorders. Biochim. Biophys. Acta 2016, 1863, 956–970. [Google Scholar] [CrossRef] [PubMed]
- Peeters, A.; Swinnen, J.V.; Van Veldhoven, P.P.; Baes, M. Hepatosteatosis in peroxisome deficient liver despite increased beta-oxidation capacity and impaired lipogenesis. Biochimie 2011, 93, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.; Poussin, C.; Velagapudi, V.R.; Empsen, C.; Joffraud, M.; Beckmann, J.S.; Geerts, A.E.; Ravussin, Y.; Ibberson, M.; Oresic, M.; et al. Peroxisomal and microsomal lipid pathways associated with resistance to hepatic steatosis and reduced pro-inflammatory state. J. Biol. Chem. 2010, 285, 31011–31023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P. Interfaces between mitochondrial dynamics and disease. Cell Calcium 2016, 60, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Convertini, P.; Menga, A.; Andria, G.; Scala, I.; Santarsiero, A.; Castiglione Morelli, M.A.; Iacobazzi, V.; Infantino, V. The contribution of the citrate pathway to oxidative stress in Down syndrome. Immunology 2016, 149, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, V.; Convertini, P.; Infantino, V.; Scarcia, P.; Todisco, S.; Palmieri, F. Statins, fibrates and retinoic acid upregulate mitochondrial acylcarnitine carrier gene expression. Biochem. Biophys. Res. Commun. 2009, 388, 643–647. [Google Scholar] [CrossRef]
- Laghezza, A.; Montanari, R.; Lavecchia, A.; Piemontese, L.; Pochetti, G.; Iacobazzi, V.; Infantino, V.; Capelli, D.; De Bellis, M.; Liantonio, A.; et al. On the metabolically active form of metaglidasen: Improved synthesis and investigation of its peculiar activity on peroxisome proliferator-activated receptors and skeletal muscles. ChemMedChem 2015, 10, 555–565. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Sanderson, L.M.; Matilainen, M.; Stienstra, R.; Carlberg, C.; de Groot, P.J.; Muller, M.; Kersten, S. Comprehensive analysis of PPARalpha-dependent regulation of hepatic lipid metabolism by expression profiling. PPAR Res. 2007, 2007, 26839. [Google Scholar] [CrossRef] [Green Version]
- van Vlies, N.; Ferdinandusse, S.; Turkenburg, M.; Wanders, R.J.; Vaz, F.M. PPAR alpha-activation results in enhanced carnitine biosynthesis and OCTN2-mediated hepatic carnitine accumulation. Biochim. Biophys. Acta 2007, 1767, 1134–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makowski, L.; Noland, R.C.; Koves, T.R.; Xing, W.; Ilkayeva, O.R.; Muehlbauer, M.J.; Stevens, R.D.; Muoio, D.M. Metabolic profiling of PPARalpha−/− mice reveals defects in carnitine and amino acid homeostasis that are partially reversed by oral carnitine supplementation. FASEB J. 2009, 23, 586–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, L.M.; de Groot, P.J.; Hooiveld, G.J.; Koppen, A.; Kalkhoven, E.; Muller, M.; Kersten, S. Effect of synthetic dietary triglycerides: A novel research paradigm for nutrigenomics. PLoS ONE 2008, 3, e1681. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772. [Google Scholar] [CrossRef]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry 2015, 80, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Marsico, M.; Santarsiero, A.; Pappalardo, I.; Convertini, P.; Chiummiento, L.; Sardone, A.; Di Noia, M.A.; Infantino, V.; Todisco, S. Mitochondria-Mediated Apoptosis of HCC Cells Triggered by Knockdown of Glutamate Dehydrogenase 1: Perspective for Its Inhibition through Quercetin and Permethylated Anigopreissin A. Biomedicines 2021, 9, 1664. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Fernández-Miranda, C.; Pérez-Carreras, M.; Colina, F.; López-Alonso, G.; Vargas, C.; Solís-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Korenblat, K.M.; Magkos, F.; McCrea, J.; Patterson, B.W.; Klein, S. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2010, 95, 2727–2735. [Google Scholar] [CrossRef]
- Davidson, M.H.; Armani, A.; McKenney, J.M.; Jacobson, T.A. Safety considerations with fibrate therapy. Am. J. Cardiol. 2007, 99, 3C–18C. [Google Scholar] [CrossRef]
- Kim, S.; Ko, K.; Park, S.; Lee, D.R.; Lee, J. Effect of Fenofibrate Medication on Renal Function. Korean J. Fam. Med. 2017, 38, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, T.A. Myopathy with statin-fibrate combination therapy: Clinical considerations. Nat. Rev. Endocrinol. 2009, 5, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Asahiyama, M.; Tanaka, T.; Yamamoto, S.; Murakami, K.; Kamiya, W.; Matsumura, Y.; Osawa, T.; Anai, M.; Fruchart, J.C.; et al. Pemafibrate, a selective PPARalpha modulator, prevents non-alcoholic steatohepatitis development without reducing the hepatic triglyceride content. Sci. Rep. 2020, 10, 7818. [Google Scholar] [CrossRef]
- Seko, Y.; Yamaguchi, K.; Umemura, A.; Yano, K.; Takahashi, A.; Okishio, S.; Kataoka, S.; Okuda, K.; Moriguchi, M.; Okanoue, T.; et al. Effect of pemafibrate on fatty acid levels and liver enzymes in non-alcoholic fatty liver disease patients with dyslipidemia: A single-arm, pilot study. Hepatol. Res. 2020, 50, 1328–1336. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Eguchi, Y.; Yoneda, M.; Imajo, K.; Tamaki, N.; Suganami, H.; Nojima, T.; Tanigawa, R.; Iizuka, M.; Iida, Y.; et al. Randomised clinical trial: Pemafibrate, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), versus placebo in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2021, 54, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- Kaul, U.; Parmar, D.; Manjunath, K.; Shah, M.; Parmar, K.; Patil, K.P.; Jaiswal, A. New dual peroxisome proliferator activated receptor agonist-Saroglitazar in diabetic dyslipidemia and non-alcoholic fatty liver disease: Integrated analysis of the real world evidence. Cardiovasc. Diabetol. 2019, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159.e5. [Google Scholar] [CrossRef] [Green Version]
- Gawrieh, S.; Noureddin, M.; Loo, N.; Mohseni, R.; Awasty, V.; Cusi, K.; Kowdley, K.V.; Lai, M.; Schiff, E.; Parmar, D.; et al. Saroglitazar, a PPAR-alpha/gamma Agonist, for Treatment of NAFLD: A Randomized Controlled Double-Blind Phase 2 Trial. Hepatology 2021, 74, 1809–1824. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.J.; Cho, Y.M.; Lee, B.W. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J. Korean Med. Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todisco, S.; Santarsiero, A.; Convertini, P.; De Stefano, G.; Gilio, M.; Iacobazzi, V.; Infantino, V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology 2022, 11, 792. https://doi.org/10.3390/biology11050792
Todisco S, Santarsiero A, Convertini P, De Stefano G, Gilio M, Iacobazzi V, Infantino V. PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology. 2022; 11(5):792. https://doi.org/10.3390/biology11050792
Chicago/Turabian StyleTodisco, Simona, Anna Santarsiero, Paolo Convertini, Giulio De Stefano, Michele Gilio, Vito Iacobazzi, and Vittoria Infantino. 2022. "PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH)" Biology 11, no. 5: 792. https://doi.org/10.3390/biology11050792
APA StyleTodisco, S., Santarsiero, A., Convertini, P., De Stefano, G., Gilio, M., Iacobazzi, V., & Infantino, V. (2022). PPAR Alpha as a Metabolic Modulator of the Liver: Role in the Pathogenesis of Nonalcoholic Steatohepatitis (NASH). Biology, 11(5), 792. https://doi.org/10.3390/biology11050792