Simple Summary
Pulmonary hypertension (PH) is a condition characterized by increased pulmonary arterial pressure. PH can further lead to right ventricular hypertrophy (RVH) and, consequently, heart failure. Endothelial-to-mesenchymal transition (EndMT) was identified as a key process in PH pathology. Nonetheless, the exact systemic and local levels of EndMT factors have not been comprehensively studied so far. Here, we quantified S100A4, epidermal growth factor (EGF), and EGF receptor (EGFR) in serum and tissue samples of two classes of PH with RVH. Patients with left heart disease and healthy volunteers served as controls. Serum S100A4 was decreased in the PH groups investigated, while EGF levels were increased in one PH cohort. EGFR was diminished in all groups compared to healthy controls. Surgical treatment of PH showed no effect on systemic EndMT marker levels. Furthermore, we observed a positive correlation between advanced PH stages and S100A4. Together, these findings help to deepen our understanding of the complex molecular events contributing to PH pathology and disease progression.
Abstract
Pulmonary hypertension (PH) is characterized by increased pulmonary arterial pressure caused by the accumulation of mesenchymal-like cells in the pulmonary vasculature. PH can lead to right ventricular hypertrophy (RVH) and, ultimately, heart failure and death. In PH etiology, endothelial-to-mesenchymal transition (EndMT) has emerged as a critical process governing the conversion of endothelial cells into mesenchymal cells, and S100A4, EGF, and EGFR are implicated in EndMT. However, a potential role of S100A4, EGF, and EGFR in PH has to date not been elucidated. We therefore quantified S100A4, EGF, and EGFR in patients suffering from chronic thromboembolic pulmonary hypertension (CTEPH) and idiopathic pulmonary arterial hypertension (iPAH). To determine specificity for unilateral heart disease, the EndMT biomarker signature was further compared between PH patients presenting with RVH and patients suffering from aortic valve stenosis (AVS) with left ventricular hypertrophy. Reduced S100A4 concentrations were found in CTEPH and iPAH patients with RVH. Systemic EGF was increased in CTEPH but not in iPAH, while AVS patients displayed slightly diminished EGF levels. EGFR was downregulated in all patient groups when compared to healthy controls. Longitudinal data analysis revealed no effect of surgical therapies on EndMT markers. Pulmonary thrombo-endarterectomized samples were devoid of S100A4, while S100A4 tissue expression positively correlated with higher grades of Heath–Edwards histopathological lesions of iPAH-derived lung tissue. Histologically, EGFR was not detectable in CTEPH lungs or in iPAH lesions. Together, our data suggest an intricate role for S100A4 and EGF/EGFR in PH with right heart pathology.
1. Introduction
The 2015 consensus guidelines of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) define pulmonary hypertension (PH) as an increased mean pulmonary arterial pressure (mPAP) ≥ 25 mmHg at rest [1,2]. Recently, the definition of PH was newly defined with an mPAP threshold > 20 mmHg [3,4]. PH may be associated with different cardiovascular and pulmonary diseases. Hence, six distinct subgroups are recognized by ESC/ERS based on different causes and conditions of PH [5]. The first group represents pulmonary arterial hypertension (PAH), which can be elicited by various conditions, such as mutations of specific loci, certain drug treatments, congenital heart disease, liver disease, human immunodeficiency virus infection, or autoimmune diseases. The exact underlying cause, however, remains obscure in the prevailing majority of PAH cases. These cases are collectively referred to as idiopathic PAH (iPAH) [6]. The incidence of PAH was determined as six cases per million [7]. Even with clinical management, five-year survival rates of patients suffering from PAH are below 60% [8]. Characteristic histopathological changes of iPAH occur in distal pulmonary arteries smaller than 500 µm in diameter, and it is postulated that dysfunctional cells of the pulmonary vasculature are responsible for these alterations. A modified Heath–Edwards classification system is commonly used to grade histopathological changes observed in patients with iPAH, which typically include intimal hyperplasia, media hypertrophy, transmural inflammation, and thrombotic and characteristic plexiform lesions [9,10]. Continuous inflammation, vasoconstriction, and obstructive vascular remodeling contribute to increased pulmonary vascular resistance (PVR), which is propagated to the heart where it manifests as pressure overload to the right ventricle [11]. Clinical management of PAH usually depends on disease severity and is based on the response to treatment. Balloon atrial septostomy (BAS) and lung transplantation (LuTX) represent the gold standard surgical interventions of PAH.
Chronic thromboembolic pulmonary hypertension (CTEPH) represents a distinct subgroup in the ESC/ERS clinical classification of PH [5]. CTEPH is often triggered by unresolved pulmonary embolism (PE), though alternative underlying mechanisms have also been identified [12]. An estimated 0.1–9.1% of patients with symptomatic PE develop CTEPH within two years after diagnosis [13], though 25% cases of CTEPH have no history of PE [14]. In 2016, the incidence of CTEPH in Germany was reported to be 5.7 cases per million [15], though CTEPH is often underdiagnosed due to the non-specific symptoms of the disease. Five-year survival rates of inoperable CTEPH are below 70%. In stark contrast to iPAH, central, proximal pulmonary arteries are subjected to pathological restructuring. Similar arteriopathy as in PAH is observed in CTEPH disease pathology, including fibrosis, blood vessel thickening, and hyperproliferation [13]. Continued occlusion and ensuing vascular remodeling are followed by an increase in PVR and eventually lead to right heart failure [12]. The surgical gold standard technique is pulmonary endarterectomy (PEA), while balloon pulmonary angioplasty will be performed if patients are considered inoperable.
In spite of the different disease etiologies, progression of both iPAH and CTEPH is characterized by increased pressure overload to the right ventricle leading to right ventricular hypertrophy (RVH). Structural cardiac remodeling eventually causes progressive organ dysfunction, and heart failure represents the leading cause of death in PH patients [16]. Various factors have been identified to play a crucial role in the conversion of compensatory RV hypertrophy into decompensated RV failure, such as inflammation, neurohormones, oxidative stress, and ischemia. In contrast to PH, aortic valve stenosis (AVS) is a pathology with mechanical stress on the left ventricle, resulting in left ventricular hypertrophy. Cardiac hypertrophy in PH and AVS thus has different etiologies and represents a disease-specific tissue response to pressure overload.
Endothelial-to-mesenchymal transition (EndMT) is a process closely resembling epithelial-to-mesenchymal transition (EMT), which has been extensively studied in cancer progression and metastasis. During embryogenesis, EndMT is crucial for the development of pulmonary arteries [17] and contributes to morphogenesis of atrioventricular valves, semilunar valves, and the septum. Postnatally, EndMT is involved in the pathogenesis of vascular calcification, organ fibrosis, and PAH [18]. In PH-associated EndMT, endothelial cells of pulmonary arterioles transit into multipotent mesenchymal progenitor cells. Morphologically, endothelial cells lose their characteristic cobblestone structure and acquire a fibroblast-like shape. This reorganization is accompanied by a downregulation of endothelial markers with concomitant upregulation of characteristic mesenchymal pathways [19]. Different molecular mechanisms have been proposed to mediate EndMT, such as TGFβR-dependent signaling via TGFβ and BMP, canonical and non-canonical Wnt signaling, TNFα and IL1β inflammatory signaling, Notch signaling, or hypoxia-induced signaling. Furthermore, epidermal growth factor (EGF) and EGF receptor (EGFR) signaling promote EndMT in endothelial cells [20]. In situ evidence for EndMT in PH has been reported previously [21]. Several transcriptional factors (TFs) are implicated in the EndMT process, such as TWIST and Snail Family Transcriptional Repressor 1 and 2 (SNAI1 and SNAI2) [22], and these TFs have already been linked to PH pathology. Twist1 phosphorylation is required for hypoxia-induced PH [23], and SNAI1 and SNAI2 upregulation was found responsible for lung endothelial cells of iPAH patients acquiring a mesenchymal phenotype [24]. These TFs are known to repress the expression of endothelial cell markers, such as vascular endothelial cadherin [25] and CD31 [26], thereby contributing to the loss of an endothelial phenotype. In spite of these previous studies, systemic and local levels of additional EndMT markers in different types of PH have not yet been comprehensively studied.
S100 Calcium Binding Protein A4 (S100A4) belongs to the S100 calcium-binding protein family and is highly expressed in the bone marrow, lymphoid tissues, and lungs. Extracellular S100A4 can exert cytokine-like functions and acts synergistically with various ligands to engage different receptors, such as receptor for advanced glycation end-products (RAGE) and epidermal growth factor receptor (EGFR) [27]. Several co-ligands of S100A4 have been reported for EGFR activation, including EGF [28], heparin-binding EGF, and amphiregulin [27]. Previously, our group reported a role for RAGE signaling in PH [29], and RAGE exacerbated PH and has been proposed as a biomarker for disease severity [30]. Though S100A4 [31] and the EGF/EGFR axis have been implicated in EndMT and EndMT was found to contribute to PH, the potential role of S100A4 and EGF/EGFR in PH so far remains elusive. To this end, we sought to investigate EndMT biomarkers in patients with PH. First, we aimed to quantify serum concentrations of the EndMT molecules S100A4, EGF, and EGFR in iPAH and CTEPH. In order to test the specificity of EndMT markers for PH with pressure overload to the right ventricle, patients with left ventricular strain due to aortic valve stenosis were selected for comparison. Secondly, we aimed to determine a potential effect of surgical therapies for iPAH, CTEPH, and AVS on serum EndMT biomarkers. Lastly, we investigated tissue expressions of S100A4 and EGFR in endarterectomized specimens of CTEPH and pulmonary parenchyma of explanted lungs.
2. Materials and Methods
2.1. Ethics Statement
This study was approved by the institutional review board on human research of the Medical University of Vienna (Vienna, Austria; ethic vote # 1805/2013) and of the University of Debrecen (Debrecen, Hungary, ethic vote # 1331/2013). This study was performed in accordance with the Declaration of Helsinki and applicable local regulations. Written informed consent was obtained from all participants.
AVS aortic valve stenosis; CTEPH chronic thromboembolic pulmonary hypertension; EGF epidermal growth factor; EGFR epidermal growth factor receptor; ELISA enzyme-linked, immunosorbent assay; IHC immunohistochemistry; iPAH idiopathic pulmonary arterial hypertension; LuTX lung transplantation; LVH left ventricular hypertrophy; PEA pulmonary endarterectomy; RVH right ventricular hypertrophy; S100A4 S100 Calcium Binding Protein A4.
2.2. Study Design and Groups
This study was designed as a prospective, longitudinal, cross-sectional cohort study performed by the Department of Thoracic Surgery, Medical University of Vienna, Vienna, Austria and the Department of Cardiac Surgery, University of Debrecen, Debrecen, Hungary, between November 2013 and November 2016 (Figure 1). We enrolled 37 patients with CTEPH undergoing PEA and 19 patients with iPAH receiving lung transplantation (LuTX) at the Department of Thoracic Surgery, Medical University of Vienna. Guidelines used for patient diagnosis were established previously [2,32]. Additionally, 15 patients suffering from severe AVS undergoing aortic valve replacement at the Department of Cardiac Surgery, University of Debrecen were included. We further recruited 39 healthy volunteers with no prior evidence of pulmonary hypertension, cardiac abnormalities, infectious diseases, or malignancies. As patient groups displayed great differences in basic demographic characteristics (age and sex), we allocated age- and sex-matched subgroups of healthy volunteers to each respective patient cohort (Table 1, Table 2 and Table 3).
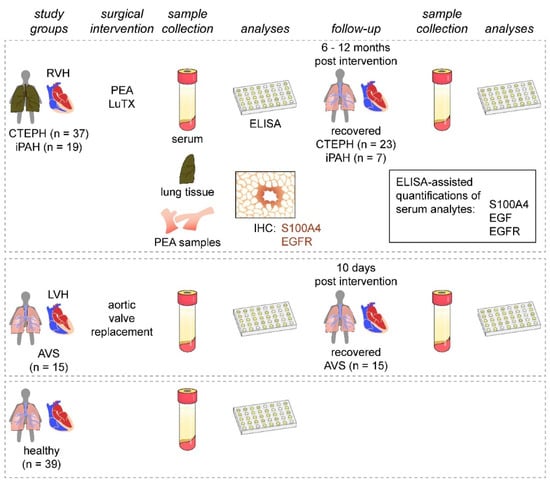
Figure 1.
Study design. Patients presenting with PH (CTEPH and iPAH) and RVH were enrolled and compared to patients with AVS and LVH. Healthy volunteers served as controls. Serum samples were obtained from all study groups and during patient follow-up visits after surgical interventions. Serum samples were analyzed for S100A4, EGF, and EGFR. Endarterectomized specimens and iPAH lung tissues were used for immunohistochemical assessment of S100A4 and EGFR expressions.

Table 1.
Basic demographic and hemodynamic data of CTEPH patients. Concentrations of EndMT biomarkers in CTEPH patients and healthy controls are reported. Controls were age- and sex-matched.
Table 2.
Basic demographic and hemodynamic data of iPAH patients. Several serum parameters of iPAH patients are shown. Concentrations of EndMT biomarkers in iPAH patients and healthy controls are reported. Controls were age- and sex-matched.
Table 3.
Basic demographic and hemodynamic data of AVS patients. Several serum parameters of AVS patients are shown. Concentrations of EndMT biomarkers in AVS patients and healthy controls are reported. Controls were age- and sex-matched.
2.3. Serum Sample Collection
Preoperative serum samples were collected one day before surgery. Postoperative serum samples of iPAH and CTEPH patients were obtained 6 to 12 months after surgical interventions. Postoperative AVS serum samples were collected 10 days after surgery. Postoperative sera were not obtained from patients with follow-up treatment at other healthcare facilities or without follow-up treatment. Healthy volunteers donated blood once (Figure 1). Blood samples were incubated at room temperature for 30 min, centrifuged at 2850 relative centrifugal force for 15 min at 4 °C, and cryopreserved <−70 °C.
2.4. Analyte Quantifications
Commercially available enzyme-linked immunosorbent assays (ELISA) were used to quantify serum S100A4, EGF, and EGFR in patient cohorts and controls (S100 Calcium Binding Protein A4—ELISA, Uscn. Life Science Inc., Aachen, Germany; Human EGF and human EGFR ELISA, both R&D Systems, Minneapolis, MN, USA). ELISAs were performed according to the manufacturers’ instructions and analyte concentrations were determined using external standards.
2.5. Tissue Sample Collection and Immunohistochemistry (IHC)
In our study, 29 cases of histologically proven iPAH and 10 CTEPH cases were histopathologically investigated by the Department of Pathology, Medical University of Vienna. Proximal specimens were collected during PEA. In parallel, samples were obtained from resected lungs of iPAH patients. Samples were formalin-fixed, paraffin-embedded, cut into serial sections of 2–5 μm thickness, and kept refrigerated until further processing.
Microsections were incubated at 50 °C for one hour and deparaffinized using 2 × Histo SAV (SAV Liquid Production GmbH, Flintsbach am Inn, Germany), propanol, ethanol (2 × 100%, 2 96%, 1 × 80%, 1 × 70%, and 1 × 50%), and distilled water. For antigen retrieval, slides were heated in a citrate buffer (Target Retrieval Solution, Dako REAL, Agilent, Santa Clara, CA, USA) (3 × 450 Watt for 5 min). Samples were subjected to 0.3% hydrogen peroxide to quench endogenous peroxidase. Slides were incubated with blocking serum (Vectastain ABC Kit, rabbit IgG, Vector Laboratories, Burlingame, CA, USA) and primary antibodies directed against human S100A4 (Abcam, rabbit polyconal IgG antibody to human S100A4, dilution: 1:200, Cambridge, UK) at 4 °C overnight. After washing, biotinylated anti-rabbit secondary antibody and avidin-peroxidase conjugates (both Vectastain) were added. Peroxidase activity was detected by 3,3′-Diaminobenzidine (DAB tablets, SIGMAFAST, Sigma Aldrich, St. Louis, MO, USA). Nuclei were visualized by Mayers’ hematoxylin. Samples were dehydrated by serial ethanol concentrations (1 × 70%, 1 × 96%, 1 × 100%), n-butyl, and xylene. Lastly, slides were mounted in resinous medium (Pertex, neutral xylene-soluble mounting medium, Medite Medizintechnik, Burgdorf, Germany).
EGFR staining was performed by autostaining using CONFIRM anti-EGFR antibody (clone 3C6, Ventana Medical Systems, Oro Valley, AZ, USA).
2.6. Statistical Methods
SPSS Statistics software version 23 (IBM SPSS Inc., Chicago, IL, USA) was used for statistical analysis. GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) was used for graphical data presentation. Normal distribution of data was determined by graphical visualization. Differences between two independent groups without normal distribution were determined by Mann–Whitney U test. Wilcoxon signed-rank test was used for longitudinal analysis of pre- and post-operative variables. Chi-square test was used to analyze independent dichotomous variables. One-way ANOVA with Bonferroni–Holm correction was used to compare three groups. p-values < 0.05 (two-tailed) were considered statistically significant and data are presented as mean ± standard deviation.
3. Results
3.1. CTEPH and iPAH Display Decreased Serum S100A4 Levels Compared to AVS and Healthy Individuals
First, we compared the pre-surgery status of serum EndMT markers between patient groups and allocated healthy controls. Before surgery, systemic S100A4 levels of CTEPH patients (1394.4 ± 960.4 pg/mL) and iPAH (1349.3 ± 894.6 pg/mL) were significantly decreased compared to healthy controls (CTEPH-control: 2065.6 ± 648.4 pg/mL; p = 0.002; iPAH-control: 2127.5 ± 706.8 pg/mL; p = 0.007) (Figure 2A,B). In contrast, S100A4 concentrations in AVS patients (2320.6 ± 727.2 pg/mL) showed no difference compared to controls (2138.5 ± 745.9 pg/mL; p = 0.465) (Figure 2C). One-way ANOVA revealed a significant downregulation of S100A4 in iPAH and CTEPH patients compared to AVS (iPAH: p = 0.008 and CTEPH: p = 0.004, respectively) (Figure 2D).
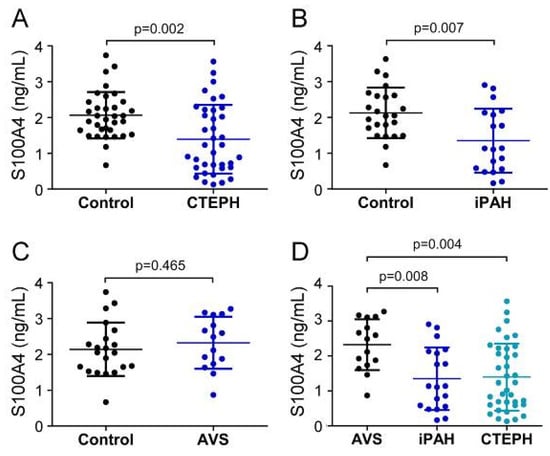
Figure 2.
Serum concentrations of S100A4 in (A) CTEPH, (B) iPAH, and (C) AVS patient groups and controls. (D) Comparison of S100A4 levels between AVS, iPAH, and CTEPH patients. Each dot represents one donor. Arithmetic means and standard deviations are indicated by horizontal lines. Patient groups were compared to controls by Mann–Whitney U test. One-way ANOVA with Bonferroni–Holm post hoc test was used to compare 3 groups.
3.2. Elevated EGF Concentration in CTEPH Compared to AVS and Controls
EGF serum concentrations in CTEPH patients (926.61 ± 576.61 pg/mL) were remarkably increased compared to controls (520.0 ± 464.2 pg/mL; p = 0.003) (Figure 3A), while EGF concentrations (766.0 ± 669.9 pg/mL) displayed no difference in the iPAH group compared to controls (511.9 ± 460.3 pg/mL; p = 0.311) (Figure 3B). Comparably, preoperative serum concentrations of EGF in the AVS group (331.3 ± 408.0 pg/mL) were not different from controls (506.5 ± 473.5 pg/mL; p = 0.400) (Figure 3C). EGF concentrations of CTEPH patients were significantly higher than the AVS group (p = 0.003) (Figure 3D). The iPAH group showed slightly yet non-significantly elevated EGF concentrations compared to the AVS group (p = 0.124).
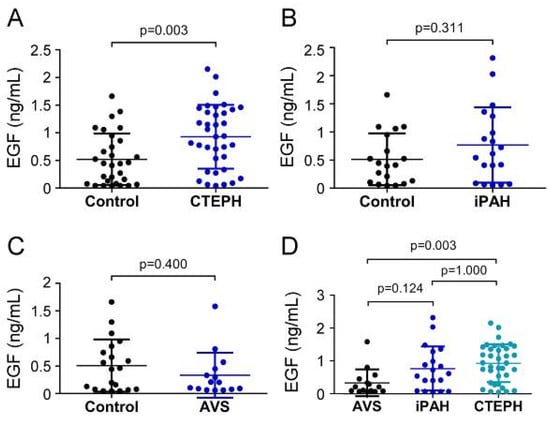
Figure 3.
Serum concentrations of EGF in (A) CTEPH, (B) iPAH, and (C) AVS patient groups and controls. (D) Comparison of EGF levels between AVS, iPAH, and CTEPH patients. Each dot represents one donor. Arithmetic means and standard deviations are indicated by horizontal lines. Patient groups were compared to controls by Mann–Whitney U test. One-way ANOVA with Bonferroni–Holm post hoc test was used to compare 3 groups.
3.3. Downregulation of Systemic EGFR Levels in CTEPH, iPAH, and AVS Compared to Controls
Analysis of EGFR serum concentrations showed that CTEPH patients (38.52 ± 15.81 ng/mL) and iPAH patients (38.6 ± 9.7 ng/mL) displayed lower values when compared to healthy volunteers (CTEPH controls 47.1 ± 11.6 ng/mL, p = 0.021; iPAH controls 49.1 ± 11.4 ng/mL, p = 0.001) (Figure 4A,B). In contrast to S100A4 and EGF, preoperative EGFR levels in AVS patients (37.4 ± 12.8 ng/mL) were decreased compared to controls (44.8 ± 9.2 ng/mL; p = 0.012) (Figure 4C). One-way ANOVA of preoperative EGFR concentrations of PH and AVS patients showed no difference (p = 0.962) (Figure 4D).
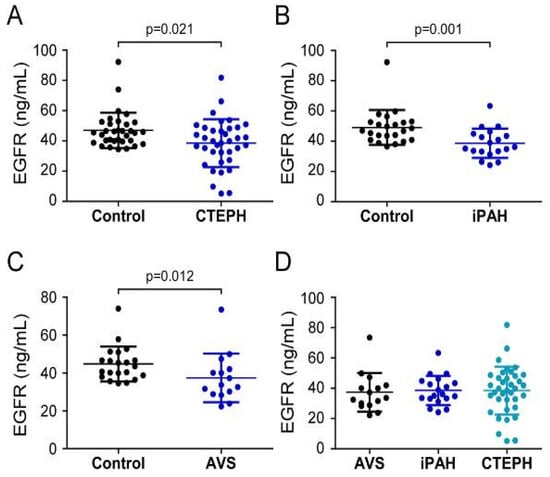
Figure 4.
Serum concentrations of EGFR in (A) CTEPH, (B) iPAH, and (C) AVS patient groups and controls. (D) Comparison of EGFR levels between AVS, iPAH, and CTEPH patients. Each dot represents one donor. Arithmetic means and standard deviations are indicated by horizontal lines. Patient groups were compared to controls by Mann–Whitney U test. One-way ANOVA with Bonferroni–Holm post hoc test was used to compare 3 groups.
3.4. Serum Analyte Concentrations Remain Unaltered by Surgical Interventions
Next, we sought to determine whether surgical therapies affected serum EndMT marker levels. Serum samples at preoperative and postoperative time points were available for 23 CTEPH, 7 iPAH, and 15 AVS patients. Patient conditions displayed pronounced improvement, evidenced by higher physical performance, reduced dyspnea, increased cardiac index, and decreased meanPAP. Longitudinal analyses revealed no difference of serum EndMT levels between pre- and post-operative time points (Figure 5).
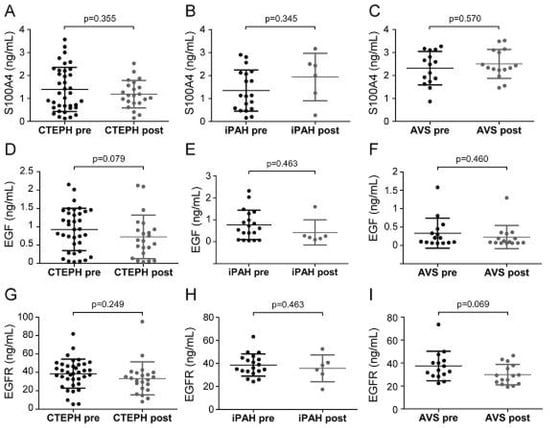
Figure 5.
Pre- and post-operative serum concentrations of S100A4, EGF, and EGFR. Pre- and post-surgical S100A4 levels were determined for (A) CTEPH, (B) iPAH, and (C) AVS. EGF concentrations before and after surgery in (D) CTEPH, (E) iPAH, and (F) AVS patients. EGFR levels of (G) CTEPH, (H) iPAH, and (I) AVS patients before surgery and after follow-up. Longitudinal comparisons were performed by Wilcoxon signed-rank test. Each dot represents one patient. Arithmetic means and standard deviations are indicated by horizontal lines.
3.5. S100A4 Expression in Lung Tissue Positively Correlates with Advanced Heath–Edwards Stages
To unravel potential sources for systemic EndMT markers, we determined tissue expression of S100A4 and EGFR in iPAH lungs and endarterectomized tissues obtained from CTEPH patients. Grade I Heath–Edwards pathohistological lesions were observed in 13 (44.8%) specimens, of which 4 (30.8%) displayed positive S100A4 staining (Figure 6A, Table 4). All (100%) higher grade lesions (≥grade II) in iPAH lungs stained positive for S100A4 (Figure 6B–D). Alveolar macrophages (Figure 6E) and epithelium (Figure 6F) expressed S100A4. Peribronchial fibrous tissue showed S100A4 staining in 96.4% of all cases (Figure 6G, Table 4), while perivascular tissue was devoid of S100A4 expression. In larger vessels, S100A4 positive intima and media were observed in 81.0% and 57.1% of all cases, respectively (Figure 6H). S100A4 and EGFR were not detectable in lung tissues of CTEPH patients (data not shown). Similarly, EGFR was not found in iPAH lesions of varying grades (Figure 7A–L). However, we observed EGFR immunoreactivity in pneumocytes of alveoli (Figure 7M) and the basal epithelium of bronchioles (Figure 7N).
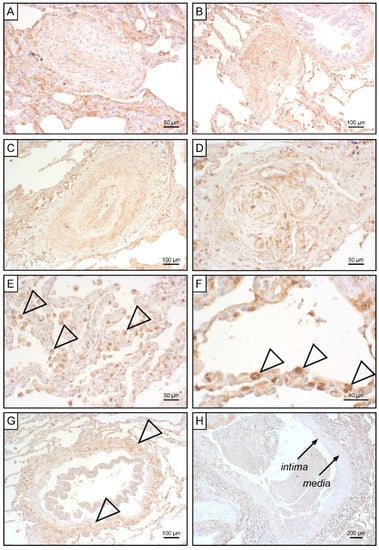
Figure 6.
S100A4 expression in iPAH patients. Immunohistochemical staining of S100A4 in iPAH lung tissues. Representative micrographs of characteristic pulmonary artery changes in small vessels according to Heath–Edwards classification (A) grade I, (B) grade II, (C) grade III, and (D) grade IV are shown. (E) Alveolar macrophages, (F) alveolar epithelium, (G) peribronchiolar fibrous tissue, and (H) tunica intima and tunica media of larger pulmonary vessels positive for S100A4 are shown. Tissues of interest are highlighted by open arrowheads.
Table 4.
S100A4 expression Heath–Edwards lesions and iPAH lungs (n = 29). Pathological vascular lesions classified according to Heath–Edwards [9,10] as well as perivascular and peribronchial expression were analyzed.
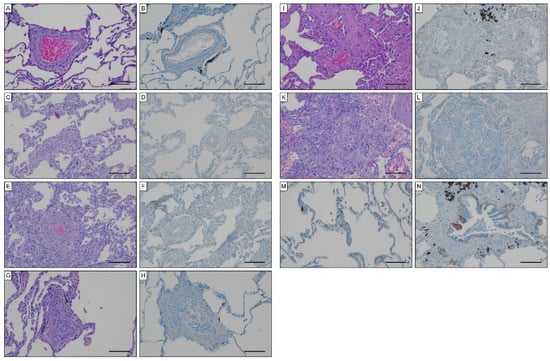
Figure 7.
EGFR expression in iPAH specimen. Hematoxylin eosin and EGFR staining in (A,B) Heath–Edwards grade I, (C,D) grade II, (E,F) grade III, (G,H) grade IV, (I,J) grade V lesions, (K,L) plexiform lesions. EGFR staining of (M) alveolar pneumocytes and (N) basal epithelium of bronchioles. Scale bars, 100 µm.
4. Discussion
In the current study, we were able to demonstrate a distinct regulation of systemic and local EndMT factors in PH with RVH. We observed a PH-specific downregulation of S100A4 and EGFR in CTEPH and in iPAH. While EGF levels of iPAH patients were comparable to controls, CTEPH displayed increased serum EGF. Intriguingly, systemic EndMT factors remained unaltered by surgical interventions. Locally, CTEPH lung specimen were devoid of S100A4 and EGFR. Lung parenchyma of iPAH patients displayed S100A4 staining in higher-grade pathological lesions, while EGFR was not detectable.
In PH pathology, substantial architectural and functional changes occur in the pulmonary vasculature. Though several underlying and perpetuating pathomechanistic events have already been identified, the picture of PH vasculopathy remains complex and incomplete. The alterations vary in different types of PH but usually involve arterial and venous restructuring. Several cell types of the lung, such as endothelial, mesenchymal, and smooth muscle cells, are involved in disease onset and progression. Originally, vascular smooth muscle cells (vSMCs) were considered the exclusive origin of alpha smooth muscle actin (αSMA)-expressing mesenchymal cells in PAH. Since then, several studies were able to demonstrate that pulmonary arteriolar endothelial cells give rise to SMC-like cells during hypoxia-induced pulmonary vascular remodeling [18,33,34]. Pulmonary endothelial cells promote inflammation and activation of adjacent SMCs resulting in pulmonary fibrosis and chronic obstruction of the affected vessels, indicating that dysregulated mesenchymal cells play a key role in the development of CTEPH [35]. In CTEPH, myofibroblast-like cells of the vascular clot induced cellular elongation and loss of cell–cell contacts in endothelial cells [36], suggesting mesenchymal cells as a contributing factor to the transition of endothelial cells. A previous work reported that endothelial cells and smooth muscle cells in pulmonary arteries of CTEPH patients obtained a migratory and proliferative phenotype in vitro [37]. These findings were extended by a study where migratory endothelial cells with a mixed mesenchymal/endothelial phenotype were detected in PAH lungs in situ [21]. An in vivo study showed that hyperproliferative, invasive myofibroblast-like cells from endarterectomized tissue of CTEPH patients were prone to develop tumors at the intimal pulmonary vessel layer in mice [38]. These data suggest that PH and the cellular microenvironment promote the conversion of healthy endothelial cells into dysfunctional mesenchymal cells, which, in turn, aggravate disease symptomatology and might serve as a potential source for systemic and local EndMT markers.
Various lung pathologies, such as plexogenic arteriopathy [39], chronic obstructive pulmonary disease [40], and several neoplastic conditions [41,42], have been associated with increased S100A4 levels and poor prognosis [43,44]. Interestingly, S100A4 has further been implicated in PH development and RVH [31], and increased S100A4 levels have been reported in the crosstalk between macrophages and pulmonary fibroblasts to promote pulmonary fibrosis [45,46]. Moreover, S100A4 is considered a crucial factor in EndMT [47,48]. Ambartsumian and colleagues reported that approximately 5% of mice ubiquitously overexpressing S100A4/Mts1 develop pulmonary artery changes resembling plexogenic lesions [49]. Intriguingly, Greenway et al. found a positive correlation between S100A4 levels and severity of vascular lesions in human plexogenic arteriopathy. The authors thus postulated that S100A4 was not implicated in the initial response to pulmonary hypertension but might exhibit functional relevance in advanced stages [39]. These data are corroborated by our histological findings where elevated S100A4 expression was detected in higher-grade pathological lesions. In detail, Greenway et al. reported S100A4 expression in neo-intimal smooth muscle cells, which is in accordance with our observation of S100A4 expression in the intima of resected iPAH lungs. Though intimal staining prevailed, we were further able to detect medial S100A4 expression. Our finding of S100A4+ alveolar macrophages is in line with data reported by Greenway et al. who described S100A4/Mts1+ periadventitial cells in mice with occlusive neointimal lesions. Together, these data indicate a role for S100A4 in PH initiation, disease progression, and the local inflammatory milieu. Delineating the exact contribution of S100A4 to the pathomechanistic events driving PH-associated vasculature remodeling merits future studies.
While the majority of previous studies was confined to investigating the local S100A4 status, we assessed S100A4 on a systemic level. Our quantifications of serum S100A4 in healthy controls are in line with a previous report of plasma S100A4 by Peng and colleagues [50]. However, Peng et al. observed strongly elevated plasma S100A4 in patients with portopulmonary hypertension, a subtype of group 1 PH with portal hypertension. In contrast to these reports, we detected decreased serum S100A4 in CTEPH and iPAH and S100A4 levels displayed a tendency towards further decrease following surgical intervention and recovery. S100A4 can be secreted and exerts paracrine, extracellular functions [51]. Interestingly, Schneider et al. observed that cardiac myocytes from patients with ischemic cardiomyopathy and aortic stenosis stained positive for S100A4 but were devoid of S100A4 mRNA [52]. The authors thus surmised that cardiomyocytes take up or bind S100A4 released from other cells rather than synthesizing S100A4 themselves. This finding might provide a potential explanation for the diminished S100A4 serum levels detected in our study groups. The mechanisms perpetuating S100A4 secretion might become exhausted in the more advanced stages of PH, the remaining S100A4 being resorbed by target tissues. Further investigations will be required to determine the origin and destination of circulating S100A4 in PH. Furthermore, S100A4 tissue levels seem to be strongly associated with disease stage. In our study, we mainly enrolled patients with end stage PH. Hence, the EndMT dynamics detected here might no longer resemble the status at disease onset or during disease progression. As EndMT might play distinct roles in disease onset, progression, and end stage, more elaborate studies are necessary to capture the dynamic turnover of systemic and local EndMT factors in the course of PH.
EGFR is overexpressed in several cancers and dysregulated EGFR signaling contributes to cancer metastasis and progression by promoting epithelial-to-mesenchymal transition [53]. Though the EGF/EGFR axis is implicated in EndMT [20], systemic EGFR was downregulated in CTEPH, in iPAH, and in AVS compared to healthy controls. Conversely, systemic EGF levels remained unaltered in iPAH and AVS but were elevated in CTEPH. Interestingly, intravenous administration of EGF induced medial thickening and increased mPAP [54]. Increased EGF levels thus might contribute to the deterioration of CTEPH pathology.
EGFR expression in pneumocytes and basal epithelial cells of the bronchioles has been shown previously [55]. In addition, only weak immunoreactivity was found in plexiform lesions of iPAH patients [55]. These data are in line with our observation of high EGFR levels in pneumocytes and the basal bronchial epithelium but no EGFR expression in pulmonary lesions. Increased expression of EGFR by pulmonary arterial smooth muscle cells was reported in PH rat models [56], and several experimental studies have already demonstrated a beneficial therapeutic effect of EGFR blockade in PH. Oral administration of an EGFR inhibitor induced regression of PAH in rats [57], subcutaneous injection of a pan EGFR inhibitor attenuated pulmonary vascular remodeling [56], and EGFR antagonists reduced RVH in experimental PAH [58]. Intriguingly, the total levels of EGFR in PH lungs were not different from healthy controls. However, EGFR auto-phosphorylation was increased in the pulmonary artery wall of patients presenting with grade 5 Heath–Edwards classification compared to controls [59]. As we observed decreased systemic EGFR levels in our groups compared to controls and no EGFR expression in the pulmonary vasculature, future studies are warranted to determine the underlying mechanisms of inhibiting EGFR to improve PH.
In order to determine whether the regulation of EndMT marker is specific for right heart disease, we compared the parameters between PH patients suffering from RVH with those of AVS patients suffering from left heart disease. Serum S100A4 levels were downregulated in PH, while showing no difference to controls in AVS. EGF was elevated in CTEPH but were not different from controls in iPAH and AVS. By comparison, EGFR was diminished in all groups investigated when compared to healthy individuals. Though several studies demonstrated a role for EGFR-dependent signaling in cardiac hypertrophy and EGFR inhibitors attenuated organ deterioration [60,61,62], our findings indicate that systemic EGF and EGFR display no specificity for PH and RVH. Of note, our data revealed that systemic downregulation of S100A4 was PH- and RVH-specific. Intriguingly, our finding of decreased systemic S100A4 is in contrast to previous reports, where elevated S100A4 tissue levels were associated with cardiac hypertrophy. S100A4 expression in cardiomyocytes of patients suffering from hypertrophic cardiomyopathy was increased [63], and S100A4 was found to be upregulated following transition to heart failure [64]. In addition, S100A4 was induced by acute ischemia and aortic stenosis and exerted pro-hypertrophic as well as cardio-protective effects [52]. These data indicate that local and systemic S100A4 in cardiac hypertrophy display opposing tendencies. Determining the potential relevance of decreased serum S100A4 in PH patients presenting with RVH remains the subject of future investigations.
Though clinical and hemodynamic parameters improved by PEA, LuTX, and AVR, pre- and post-operative serum EndMT marker levels displayed no differences. This observation is in line with our previous work where sRAGE remained unaltered by PEA, LuTX, and AVR [29]. These data indicate that surgical therapy does not affect systemic EndMT marker and that systemic levels do not reflect the prevailing disease stage.
Limitations
In spite of our best efforts, we recognize some limitations of this work. We studied a limited number of patient samples and investigating larger patient cohorts might reveal further differences and alterations in EndMT marker levels that we were not able to be captured here. Furthermore, investigating larger patient numbers allows for further patient stratification according to other characteristics, such as mortality, disease severity, and comorbidities. In our study, only operable patients were included. Patients considered not suitable to undergo surgical therapy but receiving medical therapy or rehabilitation instead were not tested. Future studies are required to compare EndMT markers between operable patients and non-surgical patients. Another limitation of our study is that patients were treated with different surgical approaches. While PEA on a cardiopulmonary bypass was performed for CTEPH patients, LuTX on extracorporeal membrane oxygenation was carried out for iPAH patients. Furthermore, the medical therapies, such as immunosuppressive agents following LuTX, differed between groups. An inevitable limitation is the reduced age of iPAH compared to CTEPH and AVS patients. We determined systemic and local EndMT marker concentrations using ELISA and IHC, respectively, and these immunoassays are not validated for clinical laboratory routine. In addition, IHC represents a qualitative rather than a quantitative method and can be subjective. Further research is necessary to elaborate on the exact tissue levels of EndMT markers. For example, quantitative PCR and Western blot analyses are more suitable to perform detailed quantification of RNA and protein levels of EndMT markers in lungs of PH patients. As our data suggest an involvement of S100A4, EGF, and EGFR in PH, the investigation of additional factors implicated in these signaling pathways will be necessary to substantiate our findings.
5. Conclusions
Taken together, our data display a heterogeneous picture of EndMT markers in PH. While S100A4 was systemically reduced in patients with PH, the tissue levels were increased in advanced grades of Heath–Edwards lesions. EGR was exclusively increased in CTEPH, but systemic EGFR was decreased in CTEPH and iPAH compared to healthy individuals. Interestingly, EGFR was not detectable in CTEPH lungs and in iPAH pathological lesions. These insights indicate a complex role of EndMT markers in PH pathology and a highly PH subtype-specific regulation of these factors. Furthermore, it seems that systemic levels do not necessarily reflect local tissue expression. This finding indicates that the disease stage cannot be determined by measuring systemic EndMT factors.
In our study, we selected the specific EndMT markers S100A4, EGF, and EGFR. Investigating further conventional EndMT markers, such as TWIST, SNAI1, SNAI2, vascular endothelial cadherin, and CD31, will be the subject of further investigations. In addition, exploring the effect of PH-specific pharmacological therapy on systemic and local EndMT markers might improve our understanding of EndMT in PH pathology. Studying EndMT factors in other cohorts, such as patients suffering from chronic thromboembolic pulmonary disease (CTEPD) with and without PH, might help to shed more light on the role of EndMT in pulmonary diseases.
Future studies are required to determine whether treatment regimens targeting processes involved in EndMT and pulmonary vascular remodeling might represent a promising therapeutic avenue in PH to prevent RVH and to decrease mortality.
6. Patents
The authors hold no patents related to this work.
Author Contributions
Conceptualization, P.H. and B.M.; methodology, P.H., F.O., J.B., T.R. and J.T.; software, P.H., F.O., J.B., T.R. and J.T.; validation, P.H.; formal analysis, P.H. and F.O.; investigation, P.H., F.O., J.B., T.R. and J.T.; resources, C.G., T.S., I.L. and N.S.-S.; data curation, M.L., P.H. and J.B.; writing—original draft preparation, M.L., P.H. and B.M.; writing—review and editing, F.O., J.B., T.R., C.G., T.S., J.T., I.L., N.S.-S. and H.J.A.; visualization, M.L. and P.H.; supervision, B.M.; project administration, B.M.; funding acquisition, H.J.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research project was supported by the Austrian Research Promotion Agency (Vienna, Austria; grant “APOSEC” 862068; 2015–2019) and the research facilities of the Department of Thoracic Surgery (FOLAB ARGE Moser and ARGE Ankersmit).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of Medical University of Vienna (protocol code 1805/2013).
Informed Consent Statement
Written informed consent was obtained from all participants.
Data Availability Statement
Raw data are available upon request.
Acknowledgments
The authors thank Heidemarie Rossiter, BSc (Hons) for linguistic proofreading.
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Delcroix, M.; Torbicki, A.; Gopalan, D.; Sitbon, O.; Klok, F.A.; Lang, I.; Jenkins, D.; Kim, N.H.; Humbert, M.; Jais, X.; et al. ERS statement on chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2021, 57, 2002828. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [CrossRef]
- Simonneau, G.; Hoeper, M.M. The revised definition of pulmonary hypertension: Exploring the impact on patient management. Eur. Heart J. Suppl. 2019, 21, K4–K8. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.M.T.; Giannoulatou, E.; Celermajer, D.S.; Humbert, M. Epidemiology and treatment of pulmonary arterial hypertension. Nat. Rev. Cardiol. 2017, 14, 603–614. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef]
- Farber, H.W.; Miller, D.P.; Poms, A.D.; Badesch, D.B.; Frost, A.E.; Muros-Le Rouzic, E.; Romero, A.J.; Benton, W.W.; Elliott, C.G.; McGoon, M.D.; et al. Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest 2015, 148, 1043–1054. [Google Scholar] [CrossRef]
- Heath, D.; Edwards, J.E. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 1958, 18, 533–547. [Google Scholar] [CrossRef]
- Carlsen, J.; Hasseriis Andersen, K.; Boesgaard, S.; Iversen, M.; Steinbruchel, D.; Bogelund Andersen, C. Pulmonary arterial lesions in explanted lungs after transplantation correlate with severity of pulmonary hypertension in chronic obstructive pulmonary disease. J. Heart Lung Transplant. 2013, 32, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Mayer, E.; Simonneau, G.; Rubin, L.J. Chronic thromboembolic pulmonary hypertension. Circulation 2006, 113, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.M.; Pesavento, R.; Bonderman, D.; Yuan, J.X. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: A current understanding. Eur. Respir. J. 2013, 41, 462–468. [Google Scholar] [CrossRef]
- Pepke-Zaba, J.; Delcroix, M.; Lang, I.; Mayer, E.; Jansa, P.; Ambroz, D.; Treacy, C.; D’Armini, A.M.; Morsolini, M.; Snijder, R.; et al. Chronic thromboembolic pulmonary hypertension (CTEPH): Results from an international prospective registry. Circulation 2011, 124, 1973–1981. [Google Scholar] [CrossRef] [PubMed]
- Kramm, T.; Wilkens, H.; Fuge, J.; Schafers, H.J.; Guth, S.; Wiedenroth, C.B.; Weingard, B.; Huscher, D.; Pittrow, D.; Cebotari, S.; et al. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clin. Res. Cardiol. 2018, 107, 548–553. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Abe, K.; Vonk Noordegraaf, A.; Voelkel, N.F. The right ventricle under pressure: Cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 2009, 135, 794–804. [Google Scholar] [CrossRef]
- Arciniegas, E.; Neves, C.Y.; Carrillo, L.M.; Zambrano, E.A.; Ramirez, R. Endothelial-mesenchymal transition occurs during embryonic pulmonary artery development. Endothelium 2005, 12, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Arciniegas, E.; Frid, M.G.; Douglas, I.S.; Stenmark, K.R. Perspectives on endothelial-to-mesenchymal transition: Potential contribution to vascular remodeling in chronic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1–L8. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Garcia de Vinuesa, A.; Ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef]
- Liu, L.; Song, S.; Zhang, Y.P.; Wang, D.; Zhou, Z.; Chen, Y.; Jin, X.; Hu, C.F.; Shen, C.X. Amphiregulin promotes cardiac fibrosis post myocardial infarction by inducing the endothelial-mesenchymal transition via the EGFR pathway in endothelial cells. Exp. Cell Res. 2020, 390, 111950. [Google Scholar] [CrossRef]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Mammoto, T.; Muyleart, M.; Konduri, G.G.; Mammoto, A. Twist1 in Hypoxia-induced Pulmonary Hypertension through Transforming Growth Factor-beta-Smad Signaling. Am. J. Respir. Cell Mol. Biol. 2018, 58, 194–207. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef]
- Dejana, E.; Tournier-Lasserve, E.; Weinstein, B.M. The control of vascular integrity by endothelial cell junctions: Molecular basis and pathological implications. Dev. Cell 2009, 16, 209–221. [Google Scholar] [CrossRef]
- Lopez, D.; Niu, G.; Huber, P.; Carter, W.B. Tumor-induced upregulation of Twist, Snail, and Slug represses the activity of the human VE-cadherin promoter. Arch. Biochem. Biophys. 2009, 482, 77–82. [Google Scholar] [CrossRef]
- Klingelhofer, J.; Moller, H.D.; Sumer, E.U.; Berg, C.H.; Poulsen, M.; Kiryushko, D.; Soroka, V.; Ambartsumian, N.; Grigorian, M.; Lukanidin, E.M. Epidermal growth factor receptor ligands as new extracellular targets for the metastasis-promoting S100A4 protein. FEBS J. 2009, 276, 5936–5948. [Google Scholar] [CrossRef]
- Cho, C.C.; Chou, R.H.; Yu, C. Amlexanox Blocks the Interaction between S100A4 and Epidermal Growth Factor and Inhibits Cell Proliferation. PLoS ONE 2016, 11, e0161663. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.; Megerle, A.; Bekos, C.; Janik, S.; Szerafin, T.; Birner, P.; Schiefer, A.I.; Mildner, M.; Lang, I.; Skoro-Sajer, N.; et al. Local and systemic RAGE axis changes in pulmonary hypertension: CTEPH and iPAH. PLoS ONE 2014, 9, e106440. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; He, Y.; Zhu, Q.; Liu, H.; Zuo, C.; Chen, G.; Yu, Y.; Lu, A. RAGE-mediated extracellular matrix proteins accumulation exacerbates HySu-induced pulmonary hypertension. Cardiovasc. Res. 2017, 113, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Merklinger, S.L.; Wagner, R.A.; Spiekerkoetter, E.; Hinek, A.; Knutsen, R.H.; Kabir, M.G.; Desai, K.; Hacker, S.; Wang, L.; Cann, G.M.; et al. Increased fibulin-5 and elastin in S100A4/Mts1 mice with pulmonary hypertension. Circ. Res. 2005, 97, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar] [CrossRef]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Huang, L.; Ge, X.; Yan, F.; Wu, R.; Ao, Q. Transdifferentiation of pulmonary arteriolar endothelial cells into smooth muscle-like cells regulated by myocardin involved in hypoxia-induced pulmonary vascular remodelling. Int. J. Exp. Pathol. 2006, 87, 463–474. [Google Scholar] [CrossRef]
- Mercier, O.; Arthur Ataam, J.; Langer, N.B.; Dorfmuller, P.; Lamrani, L.; Lecerf, F.; Decante, B.; Dartevelle, P.; Eddahibi, S.; Fadel, E. Abnormal pulmonary endothelial cells may underlie the enigmatic pathogenesis of chronic thromboembolic pulmonary hypertension. J. Heart Lung Transplant. 2017, 36, 305–314. [Google Scholar] [CrossRef]
- Sakao, S.; Hao, H.; Tanabe, N.; Kasahara, Y.; Kurosu, K.; Tatsumi, K. Endothelial-like cells in chronic thromboembolic pulmonary hypertension: Crosstalk with myofibroblast-like cells. Respir. Res. 2011, 12, 109. [Google Scholar] [CrossRef]
- Quarck, R.; Wynants, M.; Ronisz, A.; Sepulveda, M.R.; Wuytack, F.; Van Raemdonck, D.; Meyns, B.; Delcroix, M. Characterization of proximal pulmonary arterial cells from chronic thromboembolic pulmonary hypertension patients. Respir. Res. 2012, 13, 27. [Google Scholar] [CrossRef]
- Jujo, T.; Sakao, S.; Kantake, M.; Maruoka, M.; Tanabe, N.; Kasahara, Y.; Kurosu, K.; Masuda, M.; Harigaya, K.; Tatsumi, K. Characterization of sarcoma-like cells derived from endarterectomized tissues from patients with CTEPH and establishment of a mouse model of pulmonary artery intimal sarcoma. Int. J. Oncol. 2012, 41, 701–711. [Google Scholar] [CrossRef][Green Version]
- Greenway, S.; van Suylen, R.J.; Du Marchie Sarvaas, G.; Kwan, E.; Ambartsumian, N.; Lukanidin, E.; Rabinovitch, M. S100A4/Mts1 produces murine pulmonary artery changes resembling plexogenic arteriopathy and is increased in human plexogenic arteriopathy. Am. J. Pathol. 2004, 164, 253–262. [Google Scholar] [CrossRef]
- Reimann, S.; Fink, L.; Wilhelm, J.; Hoffmann, J.; Bednorz, M.; Seimetz, M.; Dessureault, I.; Troesser, R.; Ghanim, B.; Klepetko, W.; et al. Increased S100A4 expression in the vasculature of human COPD lungs and murine model of smoke-induced emphysema. Respir. Res. 2015, 16, 127. [Google Scholar] [CrossRef]
- Kimura, K.; Endo, Y.; Yonemura, Y.; Heizmann, C.W.; Schafer, B.W.; Watanabe, Y.; Sasaki, T. Clinical significance of S100A4 and E-cadherin-related adhesion molecules in non-small cell lung cancer. Int. J. Oncol. 2000, 16, 1125–1131. [Google Scholar] [CrossRef]
- Hou, S.; Tian, T.; Qi, D.; Sun, K.; Yuan, Q.; Wang, Z.; Qin, Z.; Wu, Z.; Chen, Z.; Zhang, J. S100A4 promotes lung tumor development through beta-catenin pathway-mediated autophagy inhibition. Cell Death Dis. 2018, 9, 277. [Google Scholar] [CrossRef]
- Tsuna, M.; Kageyama, S.; Fukuoka, J.; Kitano, H.; Doki, Y.; Tezuka, H.; Yasuda, H. Significance of S100A4 as a prognostic marker of lung squamous cell carcinoma. Anticancer Res. 2009, 29, 2547–2554. [Google Scholar] [PubMed]
- Miyazaki, N.; Abe, Y.; Oida, Y.; Suemizu, H.; Nishi, M.; Yamazaki, H.; Iwasaki, M.; Inoue, H.; Ueyama, Y.; Nakamura, M. Poor outcome of patients with pulmonary adenocarcinoma showing decreased E-cadherin combined with increased S100A4 expression. Int. J. Oncol. 2006, 28, 1369–1374. [Google Scholar] [CrossRef]
- Zhang, W.; Ohno, S.; Steer, B.; Klee, S.; Staab-Weijnitz, C.A.; Wagner, D.; Lehmann, M.; Stoeger, T.; Konigshoff, M.; Adler, H. S100a4 Is Secreted by Alternatively Activated Alveolar Macrophages and Promotes Activation of Lung Fibroblasts in Pulmonary Fibrosis. Front. Immunol. 2018, 9, 1216. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bao, J.; Bian, Y.; Erben, U.; Wang, P.; Song, K.; Liu, S.; Li, Z.; Gao, Z.; Qin, Z. S100A4(+) Macrophages Are Necessary for Pulmonary Fibrosis by Activating Lung Fibroblasts. Front. Immunol. 2018, 9, 1776. [Google Scholar] [CrossRef]
- Chevalier, J.; Yin, H.; Arpino, J.M.; O’Neil, C.; Nong, Z.; Gilmore, K.J.; Lee, J.J.; Prescott, E.; Hewak, M.; Rice, C.L.; et al. Obstruction of Small Arterioles in Patients with Critical Limb Ischemia due to Partial Endothelial-to-Mesenchymal Transition. iScience 2020, 23, 101251. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Sekiguchi, A.; Sagara, K.; Tanabe, H.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L.T.; Yamashita, T. Endothelial-mesenchymal transition in human atrial fibrillation. J. Cardiol. 2017, 69, 706–711. [Google Scholar] [CrossRef]
- Ambartsumian, N.; Klingelhofer, J.; Grigorian, M.; Karlstrom, O.; Sidenius, N.; Georgiev, G.; Lukanidin, E. Tissue-specific posttranscriptional downregulation of expression of the S100A4(mts1) gene in transgenic animals. Invasion Metastasis 1998, 18, 96–104. [Google Scholar] [CrossRef]
- Peng, T.; Zamanian, R.; Krowka, M.J.; Benza, R.L.; Roberts, K.E.; Taichman, D.B.; Rybak, D.; Trotter, J.F.; Brown, R.S., Jr.; Fallon, M.B.; et al. Plasma levels of S100A4 in portopulmonary hypertension. Biomarkers 2009, 14, 156–160. [Google Scholar] [CrossRef][Green Version]
- Schmidt-Hansen, B.; Klingelhofer, J.; Grum-Schwensen, B.; Christensen, A.; Andresen, S.; Kruse, C.; Hansen, T.; Ambartsumian, N.; Lukanidin, E.; Grigorian, M. Functional significance of metastasis-inducing S100A4(Mts1) in tumor-stroma interplay. J. Biol. Chem. 2004, 279, 24498–24504. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Kostin, S.; Strom, C.C.; Aplin, M.; Lyngbaek, S.; Theilade, J.; Grigorian, M.; Andersen, C.B.; Lukanidin, E.; Lerche Hansen, J.; et al. S100A4 is upregulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc. Res. 2007, 75, 40–50. [Google Scholar] [CrossRef]
- Choudhary, K.S.; Rohatgi, N.; Halldorsson, S.; Briem, E.; Gudjonsson, T.; Gudmundsson, S.; Rolfsson, O. EGFR Signal-Network Reconstruction Demonstrates Metabolic Crosstalk in EMT. PLoS Comput. Biol. 2016, 12, e1004924. [Google Scholar] [CrossRef]
- Gillespie, M.N.; Rippetoe, P.E.; Haven, C.A.; Shiao, R.T.; Orlinska, U.; Maley, B.E.; Olson, J.W. Polyamines and epidermal growth factor in monocrotaline-induced pulmonary hypertension. Am. Rev. Respir. Dis. 1989, 140, 1463–1466. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Boonstra, A.; Voskuyl, A.E.; Vonk, M.C.; Vonk-Noordegraaf, A.; van Berkel, M.P.; Mooi, W.J.; Dijkmans, B.A.; Hondema, L.S.; Smit, E.F.; et al. Platelet-derived growth factor receptor-beta and epidermal growth factor receptor in pulmonary vasculature of systemic sclerosis-associated pulmonary arterial hypertension versus idiopathic pulmonary arterial hypertension and pulmonary veno-occlusive disease: A case-control study. Arthritis Res. Ther. 2011, 13, R61. [Google Scholar] [CrossRef]
- Yu, X.; Zhao, X.; Zhang, J.; Li, Y.; Sheng, P.; Ma, C.; Zhang, L.; Hao, X.; Zheng, X.; Xing, Y.; et al. Dacomitinib, a new pan-EGFR inhibitor, is effective in attenuating pulmonary vascular remodeling and pulmonary hypertension. Eur. J. Pharmacol. 2019, 850, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Merklinger, S.L.; Jones, P.L.; Martinez, E.C.; Rabinovitch, M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation 2005, 112, 423–431. [Google Scholar] [CrossRef]
- Dahal, B.K.; Cornitescu, T.; Tretyn, A.; Pullamsetti, S.S.; Kosanovic, D.; Dumitrascu, R.; Ghofrani, H.A.; Weissmann, N.; Voswinckel, R.; Banat, G.A.; et al. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2010, 181, 158–167. [Google Scholar] [CrossRef]
- Rafikova, O.; Rafikov, R.; Kangath, A.; Qu, N.; Aggarwal, S.; Sharma, S.; Desai, J.; Fields, T.; Ludewig, B.; Yuan, J.X.; et al. Redox regulation of epidermal growth factor receptor signaling during the development of pulmonary hypertension. Free Radic. Biol. Med. 2016, 95, 96–111. [Google Scholar] [CrossRef]
- Asakura, M.; Kitakaze, M.; Takashima, S.; Liao, Y.; Ishikura, F.; Yoshinaka, T.; Ohmoto, H.; Node, K.; Yoshino, K.; Ishiguro, H.; et al. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: Metalloproteinase inhibitors as a new therapy. Nat. Med. 2002, 8, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.Y.; Chen, X.; Chen, S.R.; Li, Q.; Wang, Y.H.; Zou, J.; Cao, W.W.; Luo, J.N.; Gao, H.; Liu, P.Q. Upregulation of Nox4 promotes angiotensin II-induced epidermal growth factor receptor activation and subsequent cardiac hypertrophy by increasing ADAM17 expression. Can. J. Cardiol. 2013, 29, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Tian, X.; Qian, Y.; Skibba, M.; Zou, C.; Liu, Z.; Wang, J.; Xu, Z.; Li, X.; Liang, G. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J. Cell. Mol. Med. 2016, 20, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Han, J.; Zhao, P.; Dong, X.; Gong, S. S100A4 and P53 in myocardial collagen fibers of hypertrophic cardiomyopathy: Expression and clinical significance. Herz 2016, 41, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Iwanaga, Y.; Niizuma, S.; Kawashima, T.; Kato, T.; Inuzuka, Y.; Horie, T.; Morooka, H.; Takase, T.; Akahashi, Y.; et al. Metastasis-associated protein, S100A4 mediates cardiac fibrosis potentially through the modulation of p53 in cardiac fibroblasts. J. Mol. Cell. Cardiol. 2013, 57, 72–81. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).