
Predicting Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) of HRAS Gene and In Silico Evaluation of Their Structural and Functional Consequences towards Diagnosis and Prognosis of Cancer

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
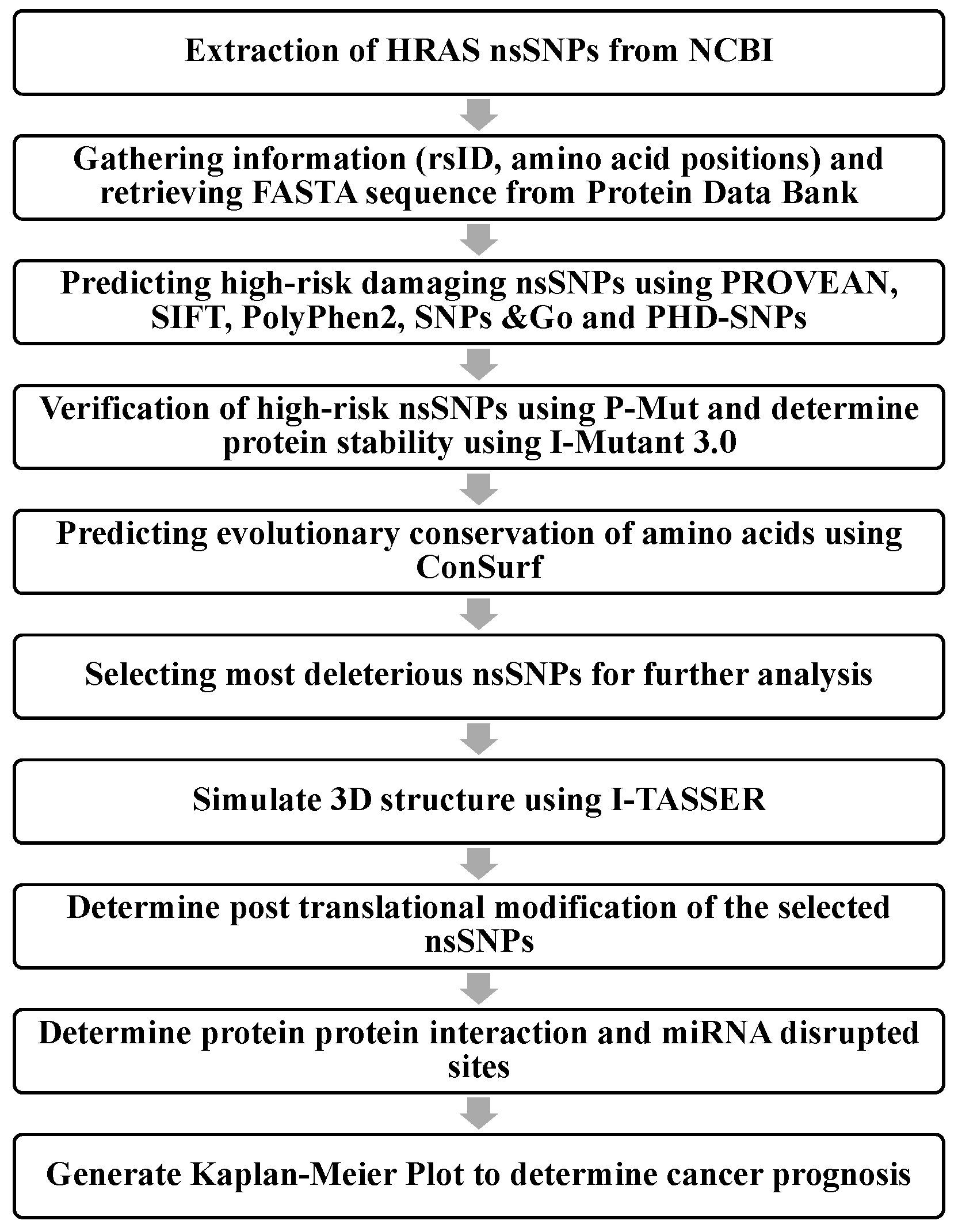
2. Materials and Methods
2.1. Retrieving nsSNPs
2.2. Identifying the Damaging nsSNPs
2.3. Verifying the High-Risk nsSNPs
2.4. Analyzing Protein Stability
2.5. Analyzing Protein Evolutionary Conservation
2.6. 3D Protein Modeling
2.7. Predicting Post-Translational Modification (PTM) Sites
2.8. Predicting Protein–Protein Interactions by Search Tool for the Retrieval of Interacting Proteins (STRING)
2.9. PolymiRTS Database 3.0
2.10. Kaplan–Meier Plotter Analysis (KM Plotter)
2.11. Molecular Dynamics Simulation
3. Results
3.1. nsSNPs Retrieved from dbSNP Database
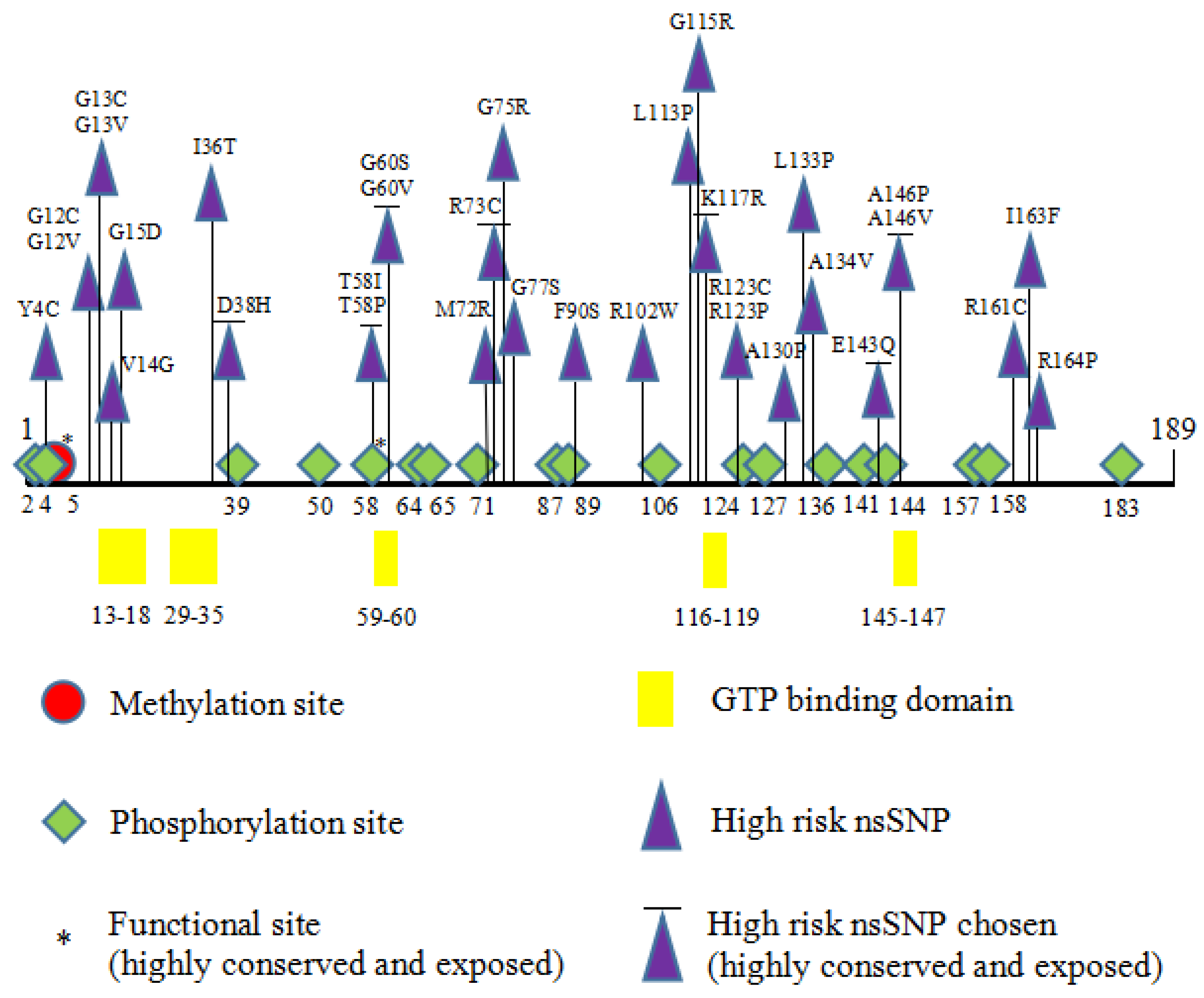
3.2. Deleterious nsSNPs Identified in HRAS Gene
3.3. Verification of 33 HRAS High-Risk nsSNPs by PMut and I-Mutant
3.4. Conservation Profile of Deleterious nsSNPs in HRAS
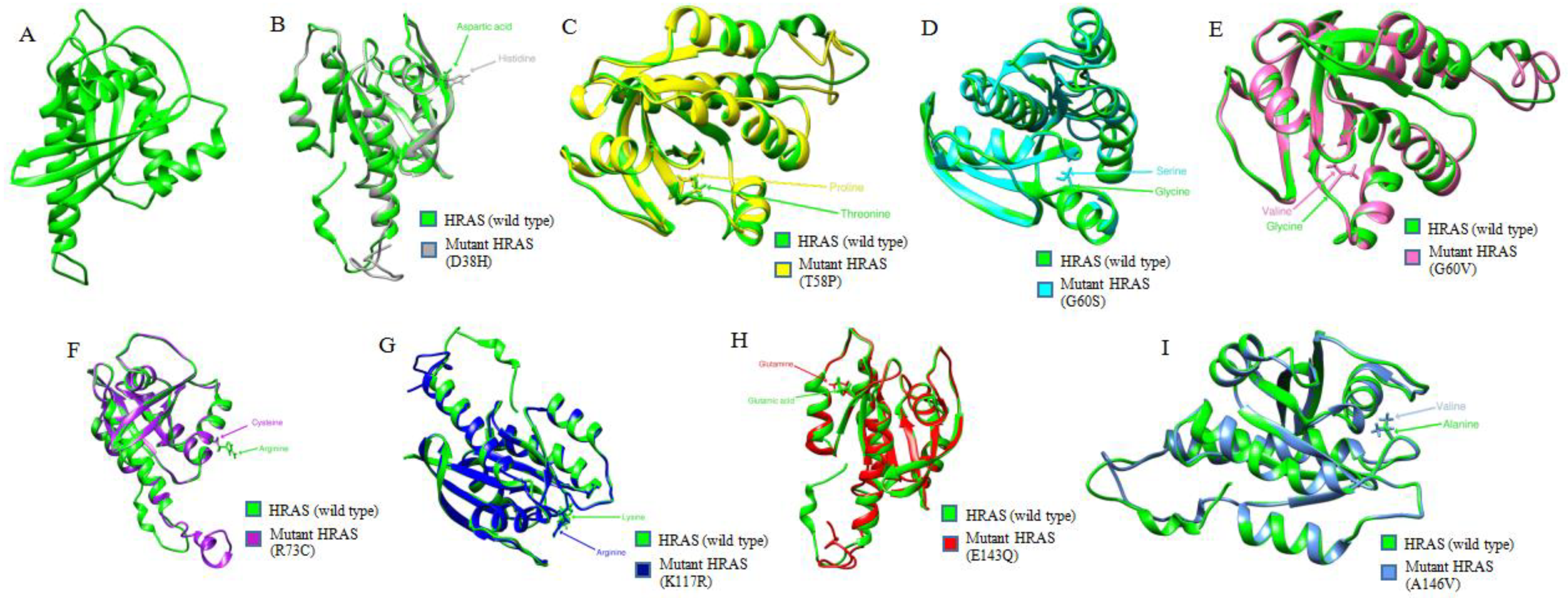
3.5. Comparative Modeling of Wild-Type HRAS and Its Mutants
3.6. Post-Translational Modifications
3.7. Protein–Protein Interaction Analysis
3.8. Prediction of nsSNPs within 3′ UTR
3.9. Expression Levels of HRAS on Overall Survival (OS) in Patients with Cancers
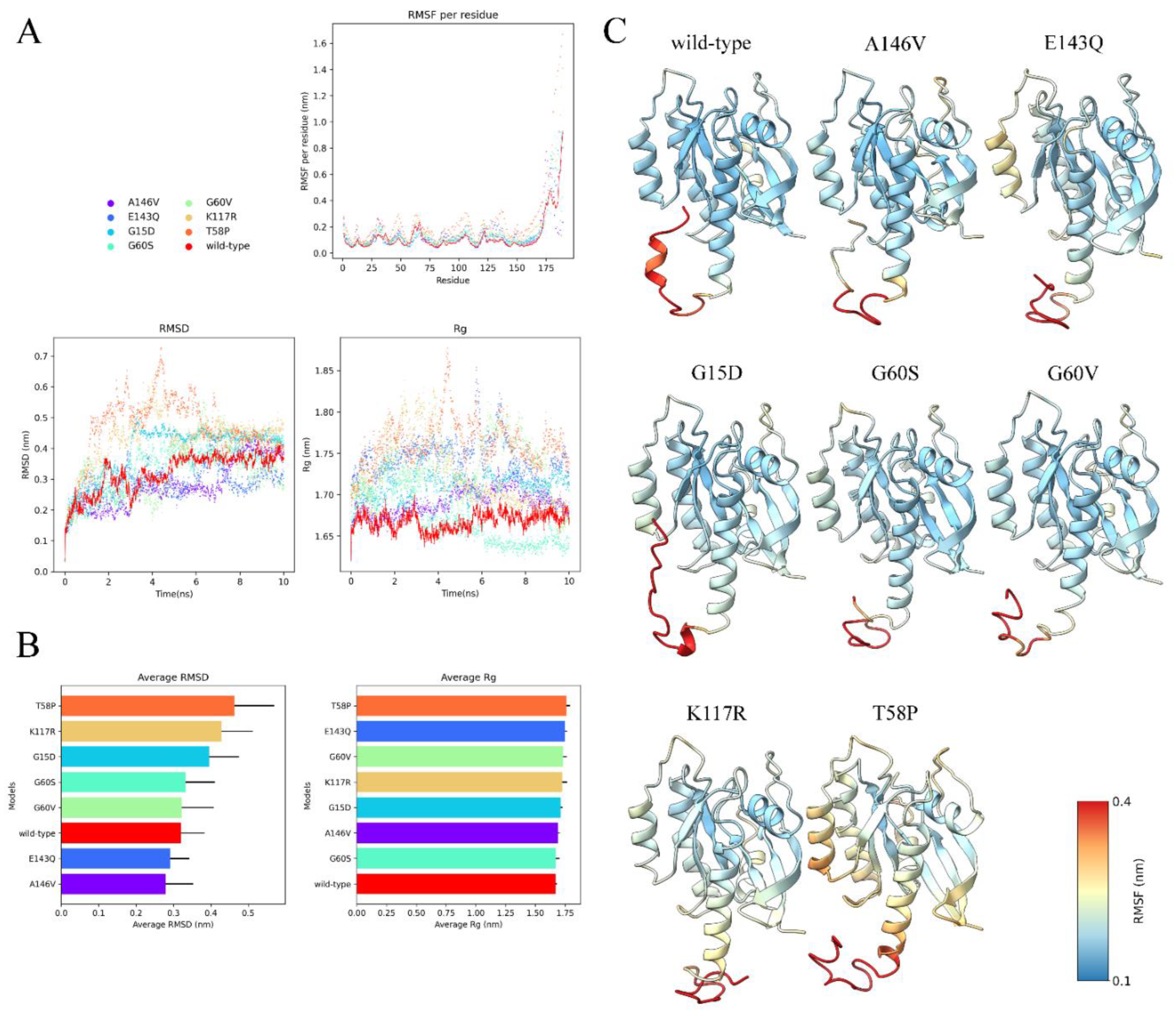
3.10. Visualization and Analysis of MD Simulation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bueno, A.; Morilla, I.; Diez, D.; Moya-Garcia, A.A.; Lozano, J.; Ranea, J.A. Exploring the interactions of the RAS family in the human protein network and their potential implications in RAS-directed therapies. Oncotarget 2016, 7, 75810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, F. A brief history of RAS and the RAS Initiative. Adv. Cancer Res. 2022, 153, 1–27. [Google Scholar] [CrossRef]
- Baines, A.T.; Xu, D.; Der, C.J. Inhibition of Ras for cancer treatment: The search continues. Future Med. Chem. 2011, 3, 1787–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhao, T.; Zhang, Y.; Zhang, K.; Shi, L.; Chen, Y.; Wang, X.; Sun, Z. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018, 46, 7793–7804. [Google Scholar] [CrossRef] [Green Version]
- Odeniyide, P.; Yohe, M.E.; Pollard, K.; Vaseva, A.V.; Calizo, A.; Zhang, L.; Rodriguez, F.J.; Gross, J.M.; Allen, A.N.; Wan, X.; et al. Targeting farnesylation as a novel therapeutic approach in HRAS-mutant rhabdomyosarcoma. Oncogene 2022, 41, 2973–2983. [Google Scholar] [CrossRef]
- Ge, W.; Goga, A.; He, Y.; Silva, P.N.; Hirt, C.K.; Herrmanns, K.; Guccini, I.; Godbersen, S.; Schwank, G.; Stoffel, M. miR-802 Suppresses Acinar-to-Ductal Reprogramming During Early Pancreatitis and Pancreatic Carcinogenesis. Gastroenterology 2022, 162, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Javaid, S.; Schaefer, A.; Goodwin, C.M.; Nguyen, V.V.; Massey, F.L.; Pierobon, M.; Gambrell-Sanders, D.; Waters, A.M.; Lambert, K.N.; Diehl, J.N.; et al. Concurrent Inhibition of ERK and Farnesyltransferase Suppresses the Growth of HRAS Mutant Head and Neck Squamous Cell Carcinoma. Mol. Cancer Ther. 2022, 21, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, D.; Yuchi, J.; He, F.; Jiang, Y.; Cai, S.; Li, J.; Xu, D. MusiteDeep: A deep-learning based webserver for protein post-translational modification site prediction and visualization. Nucleic Acids Res. 2020, 48, W140–W146. [Google Scholar] [CrossRef] [Green Version]
- Elber, R. From an SNP to a disease: A comprehensive statistical analysis. Structure 2015, 23, 1155. [Google Scholar] [CrossRef] [Green Version]
- Ponzoni, L.; Peñaherrera, D.A.; Oltvai, Z.N.; Bahar, I. Rhapsody: Predicting the pathogenicity of human missense variants. Bioinformatics 2020, 36, 3084–3092. [Google Scholar] [CrossRef]
- Zhang, X.; Wen, H.; Shi, X. Lysine methylation: Beyond histones. Acta Biochim. Biophys. Sin. 2012, 44, 14–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-function mutant p53: All the roads lead to tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, M.S.; Roy, A.S.; Islam, M.S. In silico analysis predicting effects of deleterious SNPs of human RASSF5 gene on its structure and functions. Sci. Rep. 2020, 10, 14542. [Google Scholar] [CrossRef]
- Vasuki, K.A.; Christy, H.J. Comprehensive Study of Human FBXW7 Deleterious nsSNP’s Functional Inference and Susceptibility to Gynaecological Cancer. Appl. Biochem. Biotechnol. 2022, 194, 407–433. [Google Scholar] [CrossRef] [PubMed]
- Reza, M.N.; Ferdous, N.; Emon, M.T.H.; Islam, M.S.; Mohiuddin, A.; Hossain, M.U. Pathogenic Genetic Variants From Highly Connected Cancer Susceptibility Genes Confer the Loss of Structural Stability. Sci. Rep. 2021, 11, 19264. [Google Scholar] [CrossRef] [PubMed]
- Luxembourg, B.; Mathias, D.; Körber, S.; Seifried, E. Prediction of the pathogenicity of antithrombin sequence variations by in silico methods. Thromb. Res. 2015, 135, 404–409. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- López-Ferrando, V.; Gazzo, A.; De La Cruz, X.; Orozco, M.; Gelpí, J.L. PMut: A web-based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 2017, 45, W222–W228. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Arshad, M.; Bhatti, A.; John, P. Identification and in silico analysis of functional SNPs of human TAGAP protein: A comprehensive study. PLoS ONE 2018, 13, e0188143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Wang, Y.; Ma, L.; Zhang, Y.; Ullah, S.; Xue, Y. Computational prediction of methylation types of covalently modified lysine and arginine residues in proteins. Brief. Bioinform. 2017, 18, 647–658. [Google Scholar] [CrossRef]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Zhou, J.; Zhang, Y.; Shi, Y.; Peng, D.; Xue, Y. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom. Proteom. Bioine. 2020, 18, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Gao, X.; Ren, J.; Jin, C.; Xue, Y. BDM-PUB: Computational prediction of protein ubiquitination sites with a Bayesian discriminant method. BDM-PUB Comput. Predict. Protein Ubiquitination Sites A Bayesian Discrim. Method 2009. [Google Scholar]
- Qiu, W.-R.; Xiao, X.; Lin, W.-Z.; Chou, K.-C. iUbiq-Lys: Prediction of lysine ubiquitination sites in proteins by extracting sequence evolution information via a gray system model. J. Biomol. Struct. Dyn. 2015, 33, 1731–1742. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. PolymiRTS Database 3.0: Linking polymorphisms in microRNAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014, 42, D86–D91. [Google Scholar] [CrossRef] [Green Version]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In Proceedings of the International Conference on Exascale Applications and Software, Stockholm, Sweden, 2–3 April 2014; pp. 3–27. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Kramer, A.; Venable, R.M.; Simmonett, A.C.; MacKerell Jr, A.D.; Klauda, J.B.; Pastor, R.W.; Brooks, B.R. Semi-automated optimization of the CHARMM36 lipid force field to include explicit treatment of long-range dispersion. J. Chem. Theory Comput. 2021, 17, 1562–1580. [Google Scholar] [CrossRef]
- Reimand, J.; Wagih, O.; Bader, G.D. Evolutionary constraint and disease associations of post-translational modification sites in human genomes. PLoS Genet. 2015, 11, e1004919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Dard, L.; Hubert, C.; Esteves, P.; Blanchard, W.; Bou About, G.; Baldasseroni, L.; Dumon, E.; Angelini, C.; Delourme, M.; Guyonnet-Dupérat, V.; et al. HRAS germline mutations impair LKB1/AMPK signaling and mitochondrial homeostasis in Costello syndrome models. J. Clin. Investig. 2022, 132, e131053. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Witham, S.; Takano, K.; Schwartz, C.; Alexov, E. A missense mutation in CLIC2 associated with intellectual disability is predicted by in silico modeling to affect protein stability and dynamics. Proteins Struct. Funct. Bioinform. 2011, 79, 2444–2454. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Gu, W. p53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Ting, P.Y.; Johnson, C.W.; Fang, C.; Cao, X.; Graeber, T.G.; Mattos, C.; Colicelli, J. Tyrosine phosphorylation of RAS by ABL allosterically enhances effector binding. FASEB J. 2015, 29, 3750–3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Wang, H.; Yao, X.; Zhang, D.; Xie, Y.; Cui, R.; Zhang, X. Circulating MicroRNAs in Cancer: Potential and Challenge. Front. Genet. 2019, 10, 626. [Google Scholar] [CrossRef] [Green Version]
- Donadi, E.A.; Castelli, E.C.; Arnaiz-Villena, A.; Roger, M.; Rey, D.; Moreau, P. Implications of the polymorphism of HLA-G on its function, regulation, evolution and disease association. Cell. Mol. Life Sci. 2011, 68, 369–395. [Google Scholar] [CrossRef] [Green Version]
- Bièche, I.; Coussy, F.; El-Botty, R.; Vacher, S.; Château-Joubert, S.; Dahmani, A.; Montaudon, E.; Reyes, C.; Gentien, D.; Reyal, F.; et al. HRAS is a therapeutic target in malignant chemo-resistant adenomyoepithelioma of the breast. J. Hematol. Oncol. 2021, 14, 143. [Google Scholar] [CrossRef] [PubMed]
- Zhumakayeva, A.M.; Rakhimov, K.D.; Omarova, I.M.; Arystan, L.I.; Adekenov, S.M. Experimental, Clinical and Morphological Analysis of H-Ras Oncoproteins for Locally Advanced Breast Cancer. Open Access Maced. J. Med. Sci. 2019, 7, 3153–3157. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP ID | Amino Acid Change | PROVEAN | SIFT | PolyPhen-2 | SNPs&GO | PhD-SNP | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sc | (Cutoff = −2.5) | Pred | TI | Effect | Sc | Pred | RI | Pred | RI | ||
| rs764622691 | Y4C | −6.39 | Deleterious | Damaging | 0 | Pro-damaging | 0.999 | Disease | 2 | Disease | 2 |
| rs104894229 | G12C | −7.26 | Deleterious | Damaging | 0.006 | Pos-damaging | 0.448 | Disease | 5 | Disease | 8 |
| rs104894230 | G12V | −7.21 | Deleterious | Damaging | 0.008 | Pos-damaging | 0.52 | Disease | 4 | Disease | 7 |
| rs104894228 | G13C | −7.72 | Deleterious | Damaging | 0 | Pos-damaging | 0.448 | Disease | 7 | Disease | 9 |
| rs104894226 | G13V | −7.65 | Deleterious | Damaging | 0 | Pro-damaging | 0.966 | Disease | 8 | Disease | 8 |
| rs1589793707 | V14G | −5.85 | Deleterious | Damaging | 0.001 | Pro-damaging | 1 | Disease | 2 | Disease | 7 |
| rs1554885139 | G15D | −5.66 | Deleterious | Damaging | 0.001 | Pro-damaging | 0.993 | Disease | 8 | Disease | 9 |
| rs775056058 | I36T | −3.57 | Deleterious | Damaging | 0.043 | Pro-damaging | 0.941 | Disease | 3 | Disease | 6 |
| rs750680771 | D38H | −6.02 | Deleterious | Damaging | 0 | Pro-damaging | 0.977 | Disease | 0 | Disease | 3 |
| rs121917758 | T58I | −5.82 | Deleterious | Damaging | 0 | Pro-damaging | 0.994 | Disease | 3 | Disease | 6 |
| rs770492627 | T58P | −5.82 | Deleterious | Damaging | 0.046 | Pro-damaging | 1 | Disease | 4 | Disease | 6 |
| rs1589792804 | G60S | −5.82 | Deleterious | Damaging | 0.001 | Pro-damaging | 0.959 | Disease | 4 | Disease | 6 |
| rs730880460 | G60V | −8.73 | Deleterious | Damaging | 0 | Pro-damaging | 0.997 | Disease | 5 | Disease | 7 |
| rs755488418 | M72R | −5.77 | Deleterious | Damaging | 0 | Pos-damaging | 0.873 | Disease | 2 | Disease | 7 |
| rs749674880 | R73C | −7.91 | Deleterious | Damaging | 0 | Pro-damaging | 0.97 | Disease | 5 | Disease | 9 |
| rs756190012 | G75R | −7.93 | Deleterious | Damaging | 0 | Pro-damaging | 0.999 | Disease | 5 | Disease | 8 |
| rs1309567083 | G77S | −5.94 | Deleterious | Damaging | 0.001 | Pro-damaging | 0.986 | Disease | 2 | Disease | 8 |
| rs1589792507 | F90S | −7.15 | Deleterious | Damaging | 0.006 | Pro-damaging | 0.997 | Disease | 3 | Disease | 8 |
| rs1057517913 | R102W | −6.82 | Deleterious | Damaging | 0 | Pos-damaging | 0.467 | Disease | 0 | Disease | 7 |
| rs1389645747 | L113P | −5.81 | Deleterious | Damaging | 0 | Pro-damaging | 0.986 | Disease | 5 | Disease | 8 |
| rs917210997 | G115R | −7.45 | Deleterious | Damaging | 0 | Pro-damaging | 0.99 | Disease | 5 | Disease | 7 |
| rs104894227 | K117R | −2.77 | Deleterious | Damaging | 0.004 | Pro-damaging | 0.964 | Disease | 0 | Disease | 5 |
| rs369106578 | R123C | −6.81 | Deleterious | Damaging | 0 | Pro-damaging | 0.99 | Disease | 3 | Disease | 8 |
| rs730880464 | R123P | −5.73 | Deleterious | Damaging | 0 | Pro-damaging | 1 | Disease | 6 | Disease | 8 |
| rs1564788957 | A130P | −3.27 | Deleterious | Damaging | 0.003 | Pro-damaging | 0.94 | Disease | 3 | Disease | 7 |
| rs766801436 | L133P | −4.91 | Deleterious | Damaging | 0.001 | Pro-damaging | 0.997 | Disease | 3 | Disease | 7 |
| rs397517141 | A134V | −3.49 | Deleterious | Damaging | 0.01 | Pos-damaging | 0.611 | Disease | 2 | Disease | 5 |
| rs909222512 | E143Q | −2.67 | Deleterious | Damaging | 0.009 | Pos-damaging | 0.765 | Disease | 1 | Disease | 4 |
| rs104894231 | A146P | −4.57 | Deleterious | Damaging | 0.001 | Pro-damaging | 0.994 | Disease | 6 | Disease | 7 |
| rs121917759 | A146V | −3.67 | Deleterious | Damaging | 0 | Pos-damaging | 0.596 | Disease | 4 | Disease | 7 |
| rs758956556 | R161C | −7.16 | Deleterious | Damaging | 0 | Pro-damaging | 1 | Disease | 3 | Disease | 7 |
| rs1564787934 | I163F | −3.57 | Deleterious | Damaging | 0.002 | Pro-damaging | 0.986 | Disease | 2 | Disease | 6 |
| rs753977266 | R164P | −4.66 | Deleterious | Damaging | 0.001 | Pro-damaging | 0.997 | Disease | 3 | Disease | 8 |
| nsSNP ID | Amino Acid Change | PMut | I-Mutant | TM-Align Predictions | |||
|---|---|---|---|---|---|---|---|
| Prediction | Stability | RI | DDG (kcal/mol) | TM-Score | RMSD | ||
| rs764622691 | 4, Y→ C | 0.75 (Disease) | Increase | 0 | −0.96 | 0.99193 | 0.54 |
| rs104894229 | 12, G → C | 0.79 (Disease) | Decrease | 6 | −1.20 | 0.99305 | 0.55 |
| rs104894230 | 12, G → V | 0.83 (Disease) | Decrease | 4 | −0.44 | 0.79612 | 2.03 |
| rs104894228 | 13, G → C | 0.82 (Disease) | Decrease | 3 | −1.17 | 0.99167 | 0.54 |
| rs104894226 | 13, G → V | 0.89 (Disease) | Decrease | 5 | −0.42 | 0.99167 | 0.54 |
| rs1589793707 | 14, V → G | 0.90 (Disease) | Decrease | 10 | −2.44 | 1 | 0 |
| rs1554885139 | 15, G → D | 0.90 (Disease) | Decrease | 3 | −0.80 | 0.99193 | 0.54 |
| rs775056058 | 36, I → T | 0.89 (Disease) | Decrease | 9 | −2.37 | 0.79612 | 2.03 |
| rs750680771 | 38, D → H | 0.90 (Disease) | Decrease | 5 | −0.48 | 0.98754 | 0.77 |
| rs121917758 | 58, T → I | 0.90 (Disease) | Increase | 2 | 0.19 | 0.98201 | 0.93 |
| rs770492627 | 58, T → P | 0.90 (Disease) | Decrease | 3 | −0.33 | 0.99193 | 0.54 |
| rs1589792804 | 60, G → S | 0.90 (Disease) | Decrease | 8 | −1.13 | 0.99305 | 0.55 |
| rs730880460 | 60, G → V | 0.90 (Disease) | Decrease | 6 | −0.30 | 0.99167 | 0.54 |
| rs755488418 | 72, M → R | 0.90 (Disease) | Decrease | 5 | −0.87 | 0.80282 | 1.98 |
| rs749674880 | 73, R → C | 0.89 (Disease) | Decrease | 3 | −0.99 | 0.99193 | 0.54 |
| rs756190012 | 75, G → R | 0.85 (Disease) | Decrease | 4 | −0.39 | 0.98089 | 0.92 |
| rs1309567083 | 77, G → S | 0.90 (Disease) | Decrease | 9 | −1.49 | 0.99305 | 0.55 |
| rs1589792507 | 90, F → S | 0.90 (Disease) | Decrease | 9 | −1.99 | 0.98201 | 0.93 |
| rs1057517913 | 102, R → W | 0.85 (Disease) | Decrease | 5 | −0.41 | 0.97805 | 1.05 |
| rs1389645747 | 113, L → P | 0.89 (Disease) | Decrease | 6 | −1.71 | 0.79612 | 2.03 |
| rs917210997 | 115, G → R | 0.89 (Disease) | Decrease | 3 | −0.65 | 0.99193 | 0.54 |
| rs104894227 | 117, K → R | 0.90 (Disease) | Decrease | 1 | −0.20 | 0.97805 | 1.05 |
| rs369106578 | 123, R → C | 0.83 (Disease) | Decrease | 5 | −0.78 | 0.79612 | 2.03 |
| rs730880464 | 123, R → P | 0.90 (Disease) | Decrease | 6 | −0.58 | 0.99851 | 0.2 |
| rs1564788957 | 130, A → P | 0.90 (Disease) | Decrease | 0 | −0.16 | 0.98089 | 0.92 |
| rs766801436 | 133, L → P | 0.90 (Disease) | Decrease | 5 | −1.68 | 0.98649 | 0.72 |
| rs397517141 | 134, A → V | 0.89 (Disease) | Decrease | 4 | −0.14 | 0.99857 | 0.20 |
| rs909222512 | 143, E → Q | 0.89 (Disease) | Decrease | 7 | −0.61 | 0.98929 | 0.60 |
| rs104894231 | 146, A → P | 0.90 (Disease) | Increase | 3 | −0.04 | 0.98754 | 0.77 |
| rs121917759 | 146, A → V | 0.89 (Disease) | Decrease | 2 | 0.07 | 0.98607 | 0.51 |
| rs758956556 | 161, R → C | 0.79 (Disease) | Decrease | 6 | −0.91 | 0.98459 | 0.56 |
| rs1564787934 | 163, I → F | 0.90 (Disease) | Decrease | 7 | −1.39 | 0.98754 | 0.77 |
| rs753977266 | 164, R → P | 0.80 (Disease) | Decrease | 6 | −0.75 | 0.98899 | 0.74 |
| SNP ID | Residue and Position | Conservation Score | Prediction |
|---|---|---|---|
| rs764622691 | Y4 | 5 | Buried |
| rs104894229 | G12 | 6 | Exposed |
| rs104894230 | G12 | 6 | Exposed |
| rs104894228 | G13 | 3 | Exposed |
| rs104894226 | G13 | 3 | Exposed |
| rs1589793707 | V14 | 9 | Highly conserved and exposed (f) |
| rs1554885139 | G15 | 9 | Highly conserved and buried (s) |
| rs775056058 | I36 | 8 | Buried |
| rs750680771 | D38 | 8 | Highly conserved and exposed (f) |
| rs121917758 | T58 | 9 | Highly conserved and exposed (f) |
| rs770492627 | T58 | 9 | Highly conserved and exposed (f) |
| rs1589792804 | G60 | 9 | Highly conserved and exposed (f) |
| rs730880460 | G60 | 9 | Highly conserved and exposed (f) |
| rs755488418 | M72 | 7 | Buried |
| rs749674880 | R73 | 8 | Highly conserved and exposed (f) |
| rs756190012 | G75 | 8 | Buried |
| rs1309567083 | G77 | 5 | Buried |
| rs1589792507 | F90 | 7 | Buried |
| rs1057517913 | R102 | 3 | Exposed |
| rs1389645747 | L113 | 7 | Buried |
| rs917210997 | G115 | 7 | Buried |
| rs104894227 | K117 | 9 | Highly conserved and exposed (f) |
| rs369106578 | R123 | 6 | Exposed |
| rs730880464 | R123 | 6 | Exposed |
| rs1564788957 | A130 | 2 | Buried |
| rs766801436 | L133 | 1 | Buried |
| rs397517141 | A134 | 8 | Buried |
| rs909222512 | E143 | 8 | Highly conserved and exposed (f) |
| rs104894231 | A146 | 9 | Highly conserved and exposed (f) |
| rs121917759 | A146 | 9 | Highly conserved and exposed (f) |
| rs758956556 | R161 | 3 | Exposed |
| rs1564787934 | I163 | 5 | Buried |
| rs753977266 | R164 | 3 | Exposed |
| dbSNP ID | Variant Type | miR ID | miRSite | Function Class | Context + Score Change |
|---|---|---|---|---|---|
| rs142218590 | SNP | hsa-miR-6886-5p | agCTGCGGAagct | D | −0.22 |
| hsa-miR-1184 | aGCTGCAGAagct | C | −0.386 | ||
| hsa-miR-1205 | agCTGCAGAagct | C | −0.109 | ||
| hsa-miR-1301-3p | AGCTGCAgaagct | C | −0.187 | ||
| hsa-miR-17-3p | agCTGCAGAagct | C | −0.104 | ||
| hsa-miR-3158-5p | agCTGCAGAagct | C | −0.105 | ||
| hsa-miR-4660 | AGCTGCAgaagct | C | −0.125 | ||
| hsa-miR-5047 | AGCTGCAgaagct | C | −0.187 | ||
| hsa-miR-544a | agcTGCAGAAgct | C | −0.055 | ||
| rs151229168 | SNP | hsa-miR-6886-5p | aagCTGCGGAagc | D | −0.22 |
| hsa-miR-2115-5p | aagctgTGGAAGC | C | −0.137 | ||
| hsa-miR-3692-3p | aagcTGTGGAAgc | C | −0.077 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, C.-Y.; Maran, S.; Thew, H.-Y.; Tan, Y.-C.; Rahman, N.M.A.N.A.; Cheng, W.-H.; Lai, K.-S.; Loh, J.-Y.; Yap, W.-S. Predicting Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) of HRAS Gene and In Silico Evaluation of Their Structural and Functional Consequences towards Diagnosis and Prognosis of Cancer. Biology 2022, 11, 1604. https://doi.org/10.3390/biology11111604
Chai C-Y, Maran S, Thew H-Y, Tan Y-C, Rahman NMANA, Cheng W-H, Lai K-S, Loh J-Y, Yap W-S. Predicting Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) of HRAS Gene and In Silico Evaluation of Their Structural and Functional Consequences towards Diagnosis and Prognosis of Cancer. Biology. 2022; 11(11):1604. https://doi.org/10.3390/biology11111604
Chicago/Turabian StyleChai, Chuan-Yu, Sathiya Maran, Hin-Yee Thew, Yong-Chiang Tan, Nik Mohd Afizan Nik Abd Rahman, Wan-Hee Cheng, Kok-Song Lai, Jiun-Yan Loh, and Wai-Sum Yap. 2022. "Predicting Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) of HRAS Gene and In Silico Evaluation of Their Structural and Functional Consequences towards Diagnosis and Prognosis of Cancer" Biology 11, no. 11: 1604. https://doi.org/10.3390/biology11111604
APA StyleChai, C.-Y., Maran, S., Thew, H.-Y., Tan, Y.-C., Rahman, N. M. A. N. A., Cheng, W.-H., Lai, K.-S., Loh, J.-Y., & Yap, W.-S. (2022). Predicting Deleterious Non-Synonymous Single Nucleotide Polymorphisms (nsSNPs) of HRAS Gene and In Silico Evaluation of Their Structural and Functional Consequences towards Diagnosis and Prognosis of Cancer. Biology, 11(11), 1604. https://doi.org/10.3390/biology11111604