
Oligomannose-Type Glycan Processing in the Endoplasmic Reticulum and Its Importance in Misfolding Diseases



{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
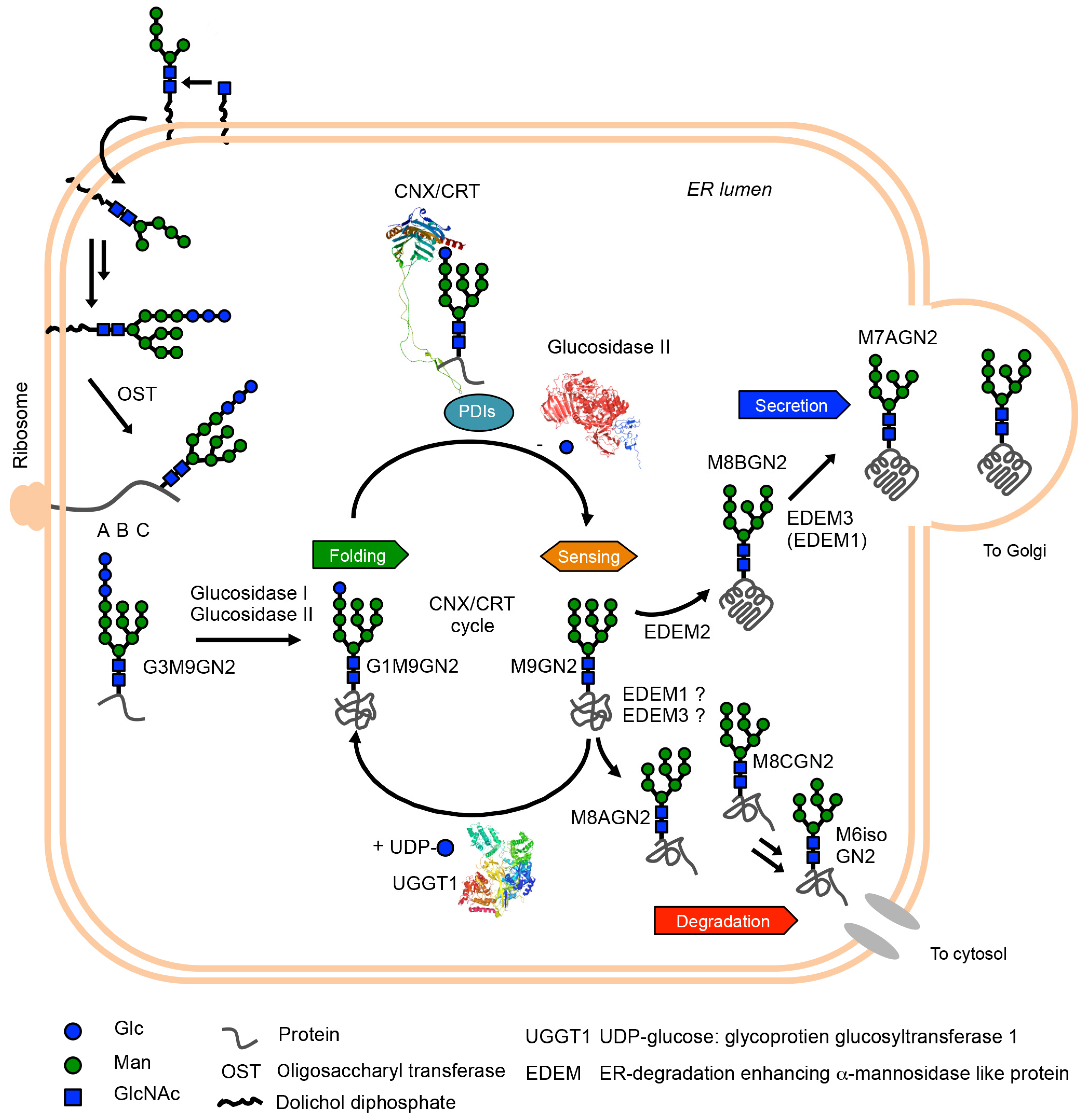
2. Glycan Roles in Glycoprotein Biosynthesis and Glycoprotein ERQC

3. Oligomannose-Type Glycan Processing in Glycoprotein ERQC and Diseases
3.1. ER Stress and Misfolding Diseases
3.1.1. Type 2 Diabetes
3.1.2. Neurodegenerative Disorders
Alzheimer’s Diseases (AD)
Parkinson’s Disease (PD)
Huntington’s Disease (HD)
Amyotrophic Lateral Sclerosis (ALS)
Prion Diseases
3.2. Oligomannose-Type Glycan Processing and Misfolding Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Chen, H.; Wang, H. Glycans of SARS-CoV-2 spike protein in virus infection and antibody production. Front. Mol. Biosci. 2021, 8, 629873. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kajihara, Y.; Takeda, Y. Chemical-synthesis-based approach to glycoprotein functions in the endoplasmic reticulum. Chem. Eur. J. 2020, 26, 15461–15470. [Google Scholar] [CrossRef] [PubMed]
- Kuribara, T.; Totani, K. Structural insights into N-linked glycan-mediated protein folding from chemical and biological perspectives. Curr. Opin. Struct. Biol. 2021, 68, 41–47. [Google Scholar] [CrossRef]
- Adams, B.M.; Oster, M.E.; Hebert, D.N. Protein quality control in the endoplasmic reticulum. Protein J. 2019, 38, 317–329. [Google Scholar] [CrossRef]
- Ninagawa, S.; George, G.; Mori, K. Mechanisms of productive folding and endoplasmic reticulum-associated degradation of glycolins and non-glycoproteins. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129812. [Google Scholar] [CrossRef]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of SWISS-PROT database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Samuelson, J.; Robbins, P.W. Effects of N-glycan precursor length diversity on quality control of protein folding and on protein glycolsylation. Semin. Cell Dev. Biol. 2015, 41, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Kelleher, D.J.; Karaoglu, D.; Mandon, E.C.; Gilmore, R. Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol. Cell 2003, 12, 101–111. [Google Scholar] [CrossRef]
- Aebi, M. N-linked protein glycolsylation in the ER. Biochim. Biophys. Acta 2013, 1833, 2430–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, Y.; Ohkawa, Y.; Kizuka, Y.; Taniguchi, N. Oligosaccharyltransferase: A gatekeeper of health and tumor progression. Int. J. Mol. Sci. 2019, 20, 6074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondruskova, N.; Cechova, A.; Hansikova, H.; Honzik, T.; Jaeken, J. Congenital disorders of glycosylation: Still “hot” in 2020. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129751. [Google Scholar] [CrossRef] [PubMed]
- Chang, I.J.; He, M.; Lam, C.T. Congenital disorders of glycosylation. Ann. Transl. Med. 2018, 6, 477. [Google Scholar] [CrossRef]
- Sun, L.; Zhao, Y.; Zhou, K.; Freeze, H.H.; Zhang, Y.W.; Xu, H. Insufficient ER–stress response caused selective mouse cerebellar granule cell degradation resembling that seen in congenital disorders of glycolsylation. Mol. Brain 2013, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, J.; Liscum, L.; Kornfeld, R. The use of 1-deoxymannojirimycin to evaluate the role of various-mannosidases in oligosaccharide processing in the intact cells. J. Biol. Chem. 1986, 261, 4766–4774. [Google Scholar] [CrossRef]
- Lu, Y.; Xu, Y.-Y.; Fan, K.-Y.; Shen, Z.-H. 1-Deoxymannojirimycin, the α1,2-mannosidase inhibitor, induced cellular endoplasmic reticulum stress in human hepatocarcinoma cell 7721. Biochem. Biophys. Res. Commun. 2006, 344, 221–225. [Google Scholar] [CrossRef]
- George, G.; Ninagawa, S.; Yagi, H.; Furukawa, J.; Hashii, N.; Ishii-Watabe, A.; Deng, Y.; Matsushita, K.; Ishikawa, T.; Mamahit, Y.P.; et al. Purified EDEM3 or EDEM1 alone produces determinant oligosaccharide structures from M8B in mammalian glycoprotein ERAD. eLife 2021, 10, e70357. [Google Scholar] [CrossRef]
- Kuribara, T.; Hirano, M.; Speciale, G.; Williams, S.J.; Ito, Y.; Totani, K. Selective manipulation of discrete mannosidase activities in the endoplasmic reticulum by using reciprocally selective inhibitors. ChemBioChem 2017, 18, 1027–1035. [Google Scholar] [CrossRef]
- Sakhi, S.; Cholet, S.; Wehbi, S.; Isidor, B.; Cogne, B.; Vuillaumier-Barrot, S.; Dupré, T.; Detleft, T.; Schmitt, E.; Leheup, B.; et al. MAN1B1-CDG: Three new individuals and associated biochemical profiles. Mol. Genet. Metab. Rep. 2021, 28, 100775. [Google Scholar] [CrossRef]
- Kamiya, Y.; Kamiya, D.; Yamamoto, K.; Nyfeler, B.; Hauri, H.-P.; Kato, K. Molecular basis of sugar recognition by the human L-type lectin ERGIC-53, VIPL, and VIP36. J. Biol. Chem. 2008, 283, 1857–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, Y.; Seko, A.; Hachisu, M.; Daikoku, S.; Izumi, M.; Koizumi, A.; Fujikawa, K.; Kajihara, Y.; Ito, Y. Both isoforms of human UDP-glucose:glycoprotein glucosyltransferase are enzymatically active. Glycobiology 2014, 24, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.T.; Alonzi, D.S.; Marti, L.; Reca, I.B.; Kiappes, J.L.; Struwe, W.B.; Cross, A.; Basu, S.; Lowe, E.D.; Darlot, B.; et al. Structures of mammalian ER alpha-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc. Natl. Acad. Sci. USA 2016, 113, E4630–E4638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blees, A.; Januliene, D.; Hofmann, T.; Koller, N.; Schmidt, C.; Trowitzsch, S.; Moeller, A.; Tampe, R. Structure of the human MHC-I peptide-loading complex. Nature 2017, 551, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Roversi, P.; Marti, L.; Caputo, A.T.; Alonzi, D.S.; Hill, J.C.; Dent, K.C.; Kumar, A.; Levasseur, M.D.; Lia, A.; Waksman, T.; et al. Interdomain conformational flexibility underpins the activity of UGGT, the eukaryotic glycoprotein secretion checkpoint. Proc. Natl. Acad. Sci. USA 2017, 114, 8544–8549. [Google Scholar] [CrossRef] [Green Version]
- Hettkamp, H.; Legler, G.; Bause, E. Purification by affinity chromatography of glucosidase I, an endoplasmic reticulum hydrolase involved in the processing of asparagine-linked oligosaccharides. Eur. J. Biochem. 1984, 142, 85–90. [Google Scholar] [CrossRef]
- Pelletier, M.F.; Marcil, A.; Sevigny, G.; Jakob, C.A.; Tessier, D.C.; Chevet, E.; Menard, R.; Bergeron, J.J.; Thomas, D.Y. The heterodimeric structure of glucosidase II is required for its activity, solubility, and localization in vivo. Glycobiology 2000, 10, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Ware, F.E.; Vassilakos, A.; Peterson, P.A.; Jackson, M.R.; Lehrman, M.A.; Williams, D.B. The molecular chaperone calnexin binds Glc1Man9GlcNAc2 oligosaccharide as an initial step in recognizing unfolded glycoproteins. J. Biol. Chem. 1995, 270, 4697–4704. [Google Scholar] [CrossRef] [Green Version]
- Michalak, M.; Milner, R.E.; Burns, K.; Opas, M. Calreticulin. Biochem. J. 1992, 28, 681–692. [Google Scholar] [CrossRef]
- Frickel, E.M.; Frei, P.; Bouvier, M.; Stafford, W.F.; Helenius, A.; Glockshuber, R.; Ellgaard, L. ERp57 is a multifunctional thiol-disulfide oxidoreductase. J. Biol. Chem. 2004, 279, 18277–18287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, H.; Seko, A.; Ito, Y.; Sakono, M. PDI family protein ERp29 recognizes P-domain of molecular chaperone calnexin. Biochem. Biophys. Res. Commun. 2017, 487, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Bastos-Aristizabal, S.; Määttänen, P.; Rosenauer, A.; Zheng, F.; Killikelly, A.; Trempe, J.F.; Thomas, D.Y.; Gehring, K. Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J. Biol. Chem. 2010, 285, 35551–35557. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Adachi, Y.; Ito, Y.; Totani, K. Calreticulin discriminates the proximal region at the N-glycosylation site of Glc1Man9GlcNAc2 ligand. Biochem. Biophys. Res. Commun. 2015, 466, 350–355. [Google Scholar] [CrossRef]
- Kozlov, G.; Gehring, K. Calnexin cycle-structural features of the ER chaperone system. FEBS J. 2020, 287, 4322–4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totani, K.; Ihara, Y.; Matsuo, I.; Ito, Y. Substrate specificity analysis of endoplasmic reticulum glucosidase II using synthetic high mannose-type glycans. J. Biol. Chem. 2006, 281, 31502–31508. [Google Scholar] [CrossRef]
- Trombetta, S.E.; Parodi, A.J. Purification to apparent homogeneity and partial characterization of rat liver UDP-glucose:glycoprotein glucosyltransferase. J. Biol. Chem. 1992, 267, 9236–9240. [Google Scholar] [CrossRef]
- Caramelo, J.J.; Castro, O.A.; Alonso, L.G.; De Prat-Gay, G.; Parodi, A.J. UDP-Glc: Glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc. Natl. Acad. Sci. USA 2003, 100, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Trombetta, E.S.; Helenius, A. Conformational requirements for glycoprotein reglucosylation in the endoplasmic reticulum. J. Cell Biol. 2000, 148, 1123–1129. [Google Scholar] [CrossRef]
- Kuribara, T.; Usui, R.; Totani, K. Glycan structure-based perspectives on the entry and release of glycoproteins in the calnexin/calreticulin cycle. Carbohydr. Res. 2021, 502, 108273. [Google Scholar] [CrossRef]
- Gonzalez, D.S.; Karaveg, K.; Vandersall-Nairn, A.S.; Lal, A.; Moremen, K.W. Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J. Biol. Chem. 1999, 274, 21375–21386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, L.O.; Herscovics, A. Cloning and expression of a specific human α1,2-mannosidase that trims Man9GlcNAc2 to Man8GlcNAc2 isomer B during N-glycan biosynthesis. Glycobiology 1999, 9, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Wada, I.; Hasegawa, K.; Yorihuzi, T.; Tremblay, L.O.; Herscovics, A.; Nagata, K. A novel ER-mannosidase-like protein accelerates ER-associated degradation. EMBO Rep. 2001, 21, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, C.A.; Burda, P.; Roth, J.; Aebi, M. Degradation of misfolded endoplasmic reticulum glycoproteins in Saccharomyces cerevisiae is determined by a specific oligosaccharide structure. J. Cell Biol. 1998, 142, 1223–1233. [Google Scholar] [CrossRef] [Green Version]
- Mast, S.W.; Diekman, K.; Karaveg, K.; Davis, A.; Sifers, R.N.; Moremen, K.W. Human EDEM2, a novel homolog of family 47 glycosidases, is involved in ER-associated degradation of glycoproteins. Glycobiology 2005, 15, 421–436. [Google Scholar] [CrossRef] [Green Version]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A.; et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Tremblay, L.O.; Sleno, B.; Kamiya, Y.; Wada, I.; Nagata, K.; Kato, K.; Herscovics, A. EDEM1 accelerates the trimming of α1,2-linked mannose on the C branch of N-glycans. Glycobiology 2010, 20, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Shenkman, M.; Ron, E.; Yehuda, R.; Benyair, R.; Khalaila, I.; Lederkremer, G.Z. Mannosidase activity of EDEM1 and EDEM2 depends on an unfolded state of their glycoprotein substrates. Commun. Biol. 2018, 1, 172. [Google Scholar] [CrossRef]
- George, G.; Ninagawa, S.; Yagi, H.; Saito, T.; Ishikawa, T.; Sakuma, T.; Yamamoto, T.; Imami, K.; Ishihama, Y.; Kato, K.; et al. EDEM2 stably disulfide-bonded to TXNDC11 catalyzes the first mannose trimming step in mammalian glycoprotein ERAD. eLife 2020, 9, e53455. [Google Scholar] [CrossRef]
- Pan, S.; Wang, S.; Utama, B.; Huang, L.; Blok, N.; Estes, M.K.; Moremen, K.W.; Sifers, R.N. Golgi localization of ERManI defines spatial separation of the mammalian glycoprotein quality control system. Mol. Biol. Cell 2011, 22, 2810–2822. [Google Scholar] [CrossRef]
- Iannotti, M.J.; Figard, L.; Sokac, A.M.; Sifers, R.N. A Golgi-localized mannosidase (MAN1B1) plays a non-enzymatic gatekeeper role in protein biosynthetic quality control. J. Biol. Chem. 2014, 289, 11844–11858. [Google Scholar] [CrossRef] [Green Version]
- Benyair, R.; Ogen-Shtern, N.; Mazkereth, N.; Shai, B.; Ehrlich, M.; Lederkremer, G.Z. Mammalian ER mannosidase I resides in quality control vesicles, where it encounters its glycoprotein substrates. Mol. Biol. Cell 2015, 26, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T.; et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, A.; Pang, P.; Kalekar, S.; Romero, P.A.; Herscovics, A.; Moremen, K.W. Substrate specificities of recombinant murine Golgi α1,2-mannosidase IA and IB and comparison with endoplasmic reticulum and Golgi processing α1,2-mannosidases. Glycobiology 1998, 8, 981–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, L.O.; Herscovics, A. Characterization of a cDNA encoding a novel human Golgi α1,2-mannosidases (IC) involved in N-glycan biosynthesis. J. Biol. Chem. 2000, 275, 31655–31660. [Google Scholar] [CrossRef] [Green Version]
- Schubert, U.; Antón, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.A.; Wilke, C.O. The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 2009, 10, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurlo, T.; Rivera, J.F.; Butler, A.E.; Cory, M.; Hoang, J.; Costes, S.; Butler, P.C. CHOP contributes to, but is not the only mediator of, IAPP induced-cell apoptosis. Mol. Endocrinol. 2016, 30, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.J.; Haataja, L.; Gurlo, T.; Butler, A.E.; Wu, X.; Soeller, W.C.; Butler, P.C. Induction of endoplasmic reticulum stress-induced-cell apoptosis and accumulation of polyubiquitinated proteins by human islet amyloid polypeptide. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1656–E1662. [Google Scholar] [CrossRef]
- Arunagiri, A.; Haataja, L.; Pottekat, A.; Pamenan, F.; Kim, S.; Zeltser, L.M.; Paton, A.W.; Paton, J.C.; Tsai, B.; Itkin-Ansari, P.; et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife 2019, 8, e44532. [Google Scholar] [CrossRef]
- Ninagawa, S.; Tada, S.; Okumura, M.; Inoguchi, K.; Kinoshita, M.; Kanemura, S.; Imami, K.; Umezawa, H.; Ishikawa, T.; Mackin, R.B.; et al. Antipsychotic olanzapine-induced misfolding of proinsulin in the endoplasmic reticulum accounts for atypical development of diabetes. eLife 2020, 9, e60970. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colla, E. Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein misfolding and ER stress in Huntington’s diseases. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Parakh, S.; Atkin, J.D. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 2016, 1648, 633–649. [Google Scholar] [CrossRef]
- May, C.E.; Soto, C. The stress of prion disease. Brain Res. 2016, 1648, 553–560. [Google Scholar]
- Costa, R.O.; Lacor, P.N.; Ferreira, I.L.; Resende, R.; Auberson, Y.P.; Klein, W.L.; Oliveira, C.R.; Rego, A.C.; Pereira, C.M.F. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-D-aspartate receptor in mature hippocampal cultures treated with amyloid-oligomers. Aging Cell 2012, 11, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Aβ and differential drug responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, T.; Doherty-Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.D.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.J.; Roy, S. α-Synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol. 2014, 24, 2319–2326. [Google Scholar] [CrossRef] [Green Version]
- Valdés, P.; Mercado, C.; Vidal, R.L.; Molina, C.; Parsons, G.; Court, F.A.; Martinez, A.; Galleguillos, D.; Armentano, D.; Schneider, B.L.; et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. USA 2014, 111, 6804–6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egawa, N.; Yamamoto, K.; Inoue, H.; Hikawa, R.; Nishi, K.; Mori, K.; Takahashi, R. The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J. Biol. Chem. 2011, 286, 7947–7957. [Google Scholar] [CrossRef] [Green Version]
- Sakahira, A.; Breuer, P.; Hayer-Hartl, M.K.; Hartl, F.U. Molecular chaperones as modulators of polyglutamine protein aggregation and toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 16412–16418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duennwald, M.L.; Lindquist, S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008, 22, 3308–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitman, J.; Ulrich Hartl, F.; Lederkremer, G.Z. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 2013, 4, 2753. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kikuchi, S.; Katada, S.; Nagai, Y.; Nishizawa, M.; Onodera, O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 2008, 17, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Iwawaki, T.; Akai, R.; Kohno, K.; Miura, M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 2004, 10, 98–102. [Google Scholar] [CrossRef]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [Green Version]
- Soo, K.Y.; Atkin, J.D.; Farg, M.; Walker, A.K.; Horne, M.K.; Nagley, P. Bim links ER stress and apoptosis in cells expressing mutant SOD1 associated with amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e35413. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Soo, K.Y.; Sundaramoorthy, V.; Parakh, S.; Ma, Y.; Farg, M.A.; Wallance, R.H.; Crouch, P.J.; Turner, B.J.; Horne, M.K.; et al. ALS-associated TDP-43 induced endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 2013, 8, e81170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farg, M.A.; Soo, K.Y.; Walker, A.K.; Pham, H.; Orian, J.; Horne, M.K.; Warraich, S.T.; Williams, K.L.; Blair, I.P.; Atkin, J.D. Mutant FUS induces endoplasmic reticulum stress in amyotrophic lateral sclerosis and interacts with protein disulphide-isomerase. Neurobiol. Aging 2012, 33, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Moumen, A.; Sunyach, C.; Pellegrino, C.; Dudley, K.; Henderson, C.E.; Raoul, C.; Pettmann, B. Reduced calreticulin levels link endoplasmic reticulum stress and Fas-triggered cell death in motoneurons vulnerable to ALS. J. Neurosci. 2012, 32, 4901–4912. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Sunyach, C.; Marissal, T.; Raoul, C.; Pettmann, B. Calreticulin levels determine onset of early muscle denervation by fast motoneurons of ALS model mice. Neurobiol. Dis. 2015, 73, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [Green Version]
- Mereno, J.A.; Radford, H.; Peretti, D.; Sreinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Mereno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef]
- Halliday, M.; Radford, H.; Sekine, Y.; Mereno, J.; Verity, N.; le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Castilla, J.; Soto, C. Perturbation of endoplasmic reticulum homeostasis facilitates prion replication. J. Biol. Chem. 2007, 282, 12725–12733. [Google Scholar] [CrossRef] [Green Version]
- Yedidia, Y.; Horonchik, L.; Tzaban, S.; Yanai, A.; Taraboulos, A. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 2001, 20, 5383–5391. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, S.; Isoyama, M.; Hirano, M.; Yamaya, K.; Ito, Y.; Matsuo, I.; Totani, K. Reconstructed glycan profile for evaluation of operating status of the endoplasmic reticulum glycoprotein quality control. Glycobiology 2013, 23, 121–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totani, K.; Ihara, Y.; Tsujimoto, T.; Matsuo, I.; Ito, Y. Recognition motif of the glycoprotein-folding sensor enzyme UDP-Glc: Glycoprotein glucosyltransferase. Biochemistry 2009, 48, 2933–2940. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Imagawa, A.; Totani, K. Stratified analysis of lectin-like chaperones in the folding disease-related metabolic syndrome rat model. Biochem. Biophys. Res. Commun. 2016, 478, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Kuribara, T.; Imagawa, A.; Hirano, M.; Ito, Y.; Totani, K. Metabolic syndrome perturbs deglucosylation and reglucosylation in the glycoprotein folding cycle. FEBS Lett. 2020, 594, 1759–1769. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuribara, T.; Totani, K. Oligomannose-Type Glycan Processing in the Endoplasmic Reticulum and Its Importance in Misfolding Diseases. Biology 2022, 11, 199. https://doi.org/10.3390/biology11020199
Kuribara T, Totani K. Oligomannose-Type Glycan Processing in the Endoplasmic Reticulum and Its Importance in Misfolding Diseases. Biology. 2022; 11(2):199. https://doi.org/10.3390/biology11020199
Chicago/Turabian StyleKuribara, Taiki, and Kiichiro Totani. 2022. "Oligomannose-Type Glycan Processing in the Endoplasmic Reticulum and Its Importance in Misfolding Diseases" Biology 11, no. 2: 199. https://doi.org/10.3390/biology11020199
APA StyleKuribara, T., & Totani, K. (2022). Oligomannose-Type Glycan Processing in the Endoplasmic Reticulum and Its Importance in Misfolding Diseases. Biology, 11(2), 199. https://doi.org/10.3390/biology11020199