
Computational Insights into the Unfolding of a Destabilized Superoxide Dismutase 1 Mutant


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. REST2 Simulations
2.2. Alchemical Calculations
3. Results
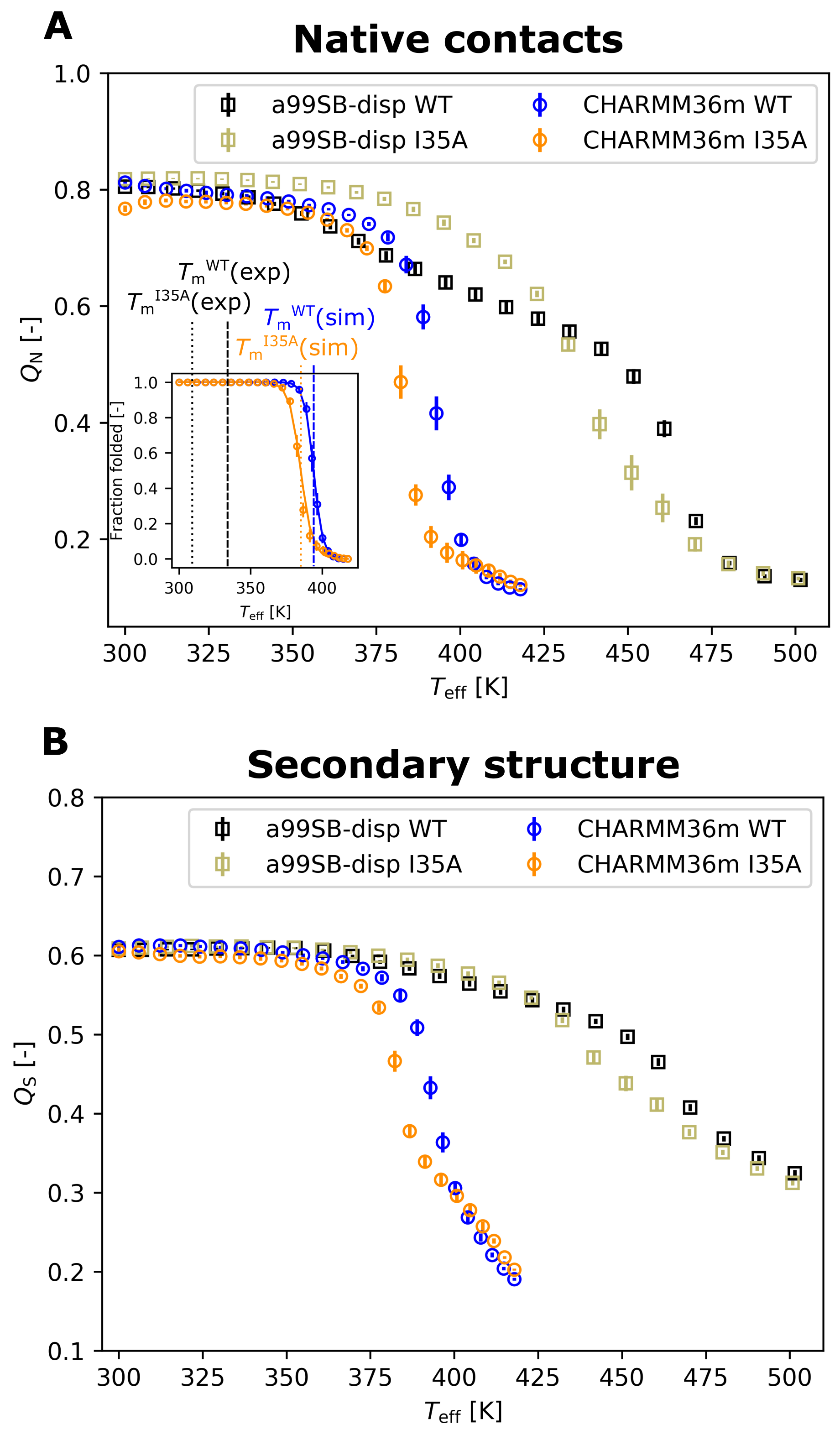
3.1. Thermal Stability from REST2 Simulations
3.2. Energetics of the Mutation
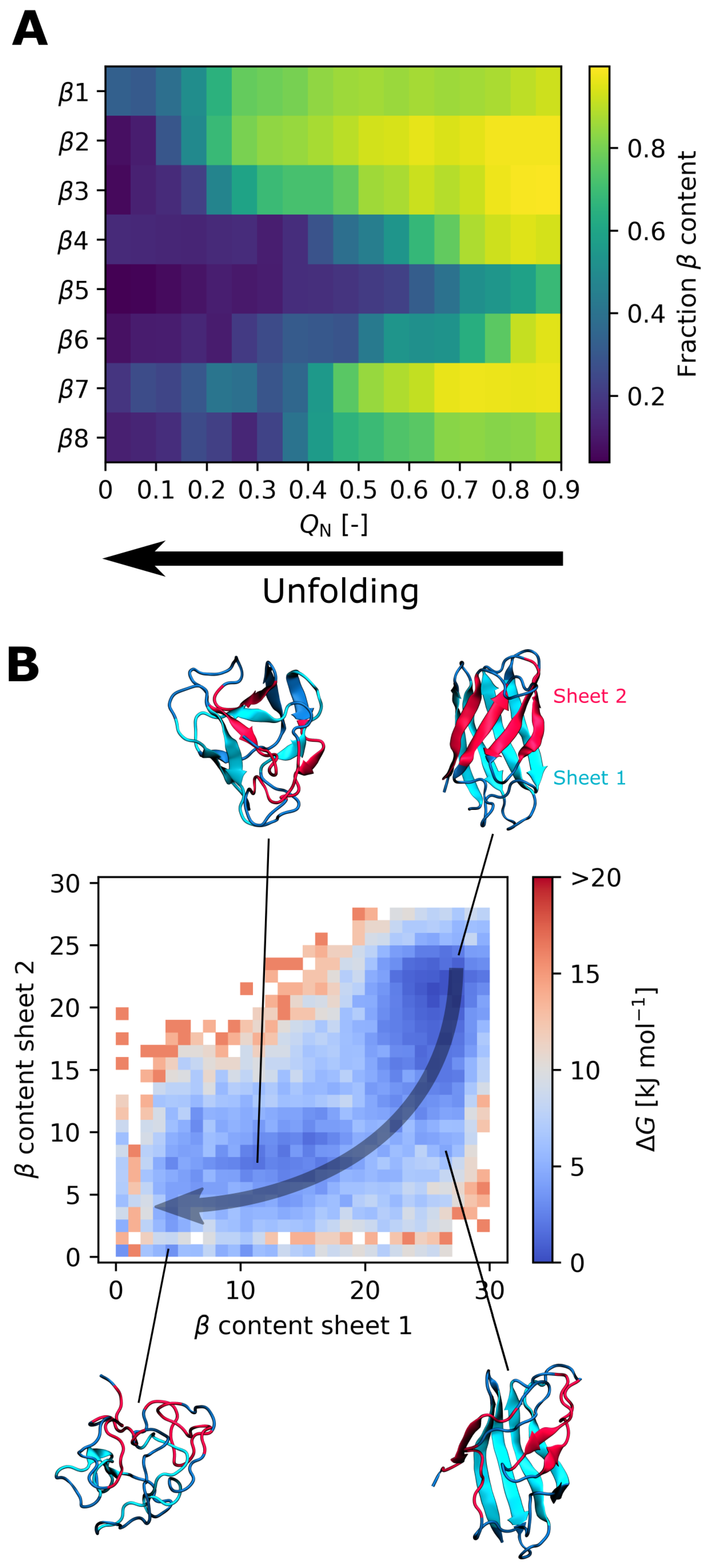
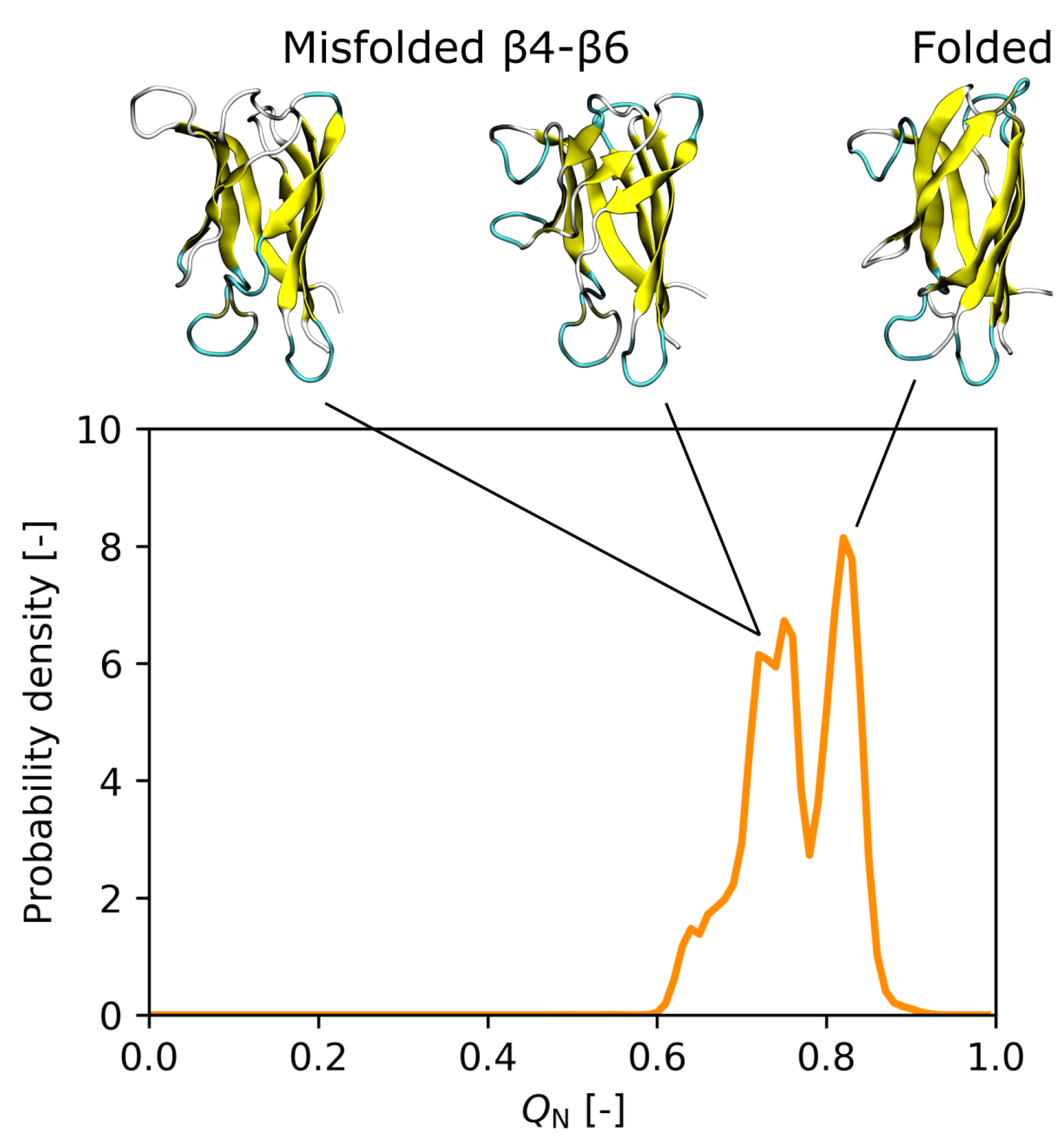
3.3. Effect of the I35A Mutation on the Folding Landscape
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Robertson, A.D.; Murphy, K.P. Protein Structure and the Energetics of Protein Stability. Chem. Rev. 1997, 97, 1251–1268. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.J.; Qu, B.H.; Pedersen, P.L. Defective protein folding as a basis of human disease. Trends Biochem. Sci. 1995, 20, 456–459. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.E.; Dokholyan, N.V.; de Simone, A.; et al. Amyloid oligomers: A joint experimental/computational perspective on Alzheimer’s disease, Parkinson’s disease, type II diabetes, and amyotrophic lateral sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Pasinelli, P.; Brown, R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat. Rev. Neurosci. 2006, 7, 710–723. [Google Scholar] [CrossRef]
- Mulligan, V.K.; Chakrabartty, A. Protein misfolding in the late-onset neurodegenerative diseases: Common themes and the unique case of amyotrophic lateral sclerosis. Proteins 2013, 81, 1285–1303. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.D.; Caplow, M.; Dokholyan, N.V. The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 15094–15099. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, A.; Rumfeldt, J.A.O.; Broom, H.R.; Doyle, C.M.; Sobering, R.E.; Meiering, E.M.; Kay, L.E. Probing the free energy landscapes of ALS disease mutants of SOD1 by NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2016, 113, E6939–E6945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Beck, M.V.; Griffith, J.D.; Deshmukh, M.; Dokholyan, N.V. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 4661–4665. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, J.; Mu, X.; Lang, L.; Wang, H.; Binolfi, A.; Theillet, F.X.; Bekei, B.; Logan, D.T.; Selenko, P.; Wennerström, H.; et al. Thermodynamics of protein destabilization in live cells. Proc. Natl. Acad. Sci. USA 2015, 112, 12402–12407. [Google Scholar] [CrossRef] [Green Version]
- Gnutt, D.; Timr, S.; Ahlers, J.; Ko, B.; Manderfeld, E.; Heyden, M.; Sterpone, F.; Ebbinghaus, S. Stability Effect of Quinary Interactions Reversed by Single Point Mutations. J. Am. Chem. Soc. 2019, 141, 4660–4669. [Google Scholar] [CrossRef] [PubMed]
- Zeineddine, R.; Farrawell, N.E.; Lambert-Smith, I.A.; Yerbury, J.J. Addition of exogenous SOD1 aggregates causes TDP-43 mislocalisation and aggregation. Cell Stress Chaperones 2017, 22, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, J.; Kurnik, M.; Lang, L.; Oliveberg, M. Cutting off functional loops from homodimeric enzyme superoxide dismutase 1 (SOD1) leaves monomeric beta-barrels. J. Biol. Chem. 2011, 286, 33070–33083. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, J.; Awad, W.; Saraboji, K.; Kurnik, M.; Lang, L.; Leinartaite, L.; Marklund, S.L.; Logan, D.T.; Oliveberg, M. Global structural motions from the strain of a single hydrogen bond. Proc. Natl. Acad. Sci. USA 2013, 110, 3829–3834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, N.; Ribeiro, S.S.; Becker, M.; Laborie, E.; Pollak, R.; Timr, S.; Sterpone, F.; Ebbinghaus, S. Sequestration of Proteins in Stress Granules Relies on the In-Cell but Not the In Vitro Folding Stability. J. Am. Chem. Soc. 2021. [Google Scholar] [CrossRef]
- Danielsson, J.; Inomata, K.; Murayama, S.; Tochio, H.; Lang, L.; Shirakawa, M.; Oliveberg, M. Pruning the ALS-Associated Protein SOD1 for in-Cell NMR. J. Am. Chem. Soc. 2013, 135, 10266–10269. [Google Scholar] [CrossRef]
- Iwakawa, N.; Morimoto, D.; Walinda, E.; Leeb, S.; Shirakawa, M.; Danielsson, J.; Sugase, K. Transient diffusive interactions with a protein crowder affect aggregation processes of superoxide dismutase 1 β-barrel. J. Phys. Chem. 2021, 125, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Sörensen, T.; Leeb, S.; Danielsson, J.; Oliveberg, M. Polyanions Cause Protein Destabilization Similar to That in Live Cells. Biochemistry 2021, 60, 735–746. [Google Scholar] [CrossRef]
- Khare, S.D.; Dokholyan, N.V. Common dynamical signatures of familial amyotrophic lateral sclerosis-associated structurally diverse Cu, Zn superoxide dismutase mutants. Proc. Natl. Acad. Sci. USA 2006, 103, 3147–3152. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Tsao, D.; Nie, H.; Dokholyan, N.V. Ab Initio Folding of Proteins with All-Atom Discrete Molecular Dynamics. Structure 2008, 16, 1010–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, E.A.; Ding, F.; Dokholyan, N.V. Structural and Thermodynamic Effects of Post-translational Modifications in Mutant and Wild Type Cu, Zn Superoxide Dismutase. J. Mol. Biol. 2011, 408, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Furukawa, Y.; Nukina, N.; Dokholyan, N.V. Local Unfolding of Cu, Zn Superoxide Dismutase Monomer Determines the Morphology of Fibrillar Aggregates. J. Mol. Biol. 2012, 421, 548–560. [Google Scholar] [CrossRef] [Green Version]
- Habibi, M.; Rottler, J.; Plotkin, S.S. The unfolding mechanism of monomeric mutant SOD1 by simulated force spectroscopy. BBA- Proteins Proteom. 2017, 1865, 1631–1642. [Google Scholar] [CrossRef]
- Peng, X.; Cashman, N.R.; Plotkin, S.S. Prediction of Misfolding-Specific Epitopes in SOD1 Using Collective Coordinates. J. Phys. Chem. 2018, 122, 11662–11676. [Google Scholar] [CrossRef] [PubMed]
- Bille, A.; Jensen, K.S.; Mohanty, S.; Akke, M.; Irbäck, A. Stability and Local Unfolding of SOD1 in the Presence of Protein Crowders. J. Phys. Chem. 2019, 123, 1920–1930. [Google Scholar] [CrossRef] [PubMed]
- Mouro, P.R.; Povinelli, A.P.; Leite, V.B.; Chahine, J. Exploring Folding Aspects of Monomeric Superoxide Dismutase. J. Phys. Chem. 2020, 124, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Timr, S.; Gnutt, D.; Ebbinghaus, S.; Sterpone, F. The Unfolding Journey of Superoxide Dismutase 1 Barrels under Crowding: Atomistic Simulations Shed Light on Intermediate States and Their Interactions with Crowders. J. Phys. Chem. Lett. 2020, 11, 4206–4212. [Google Scholar] [CrossRef]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica Exchange with Solute Scaling: A More Efficient Version of Replica Exchange with Solute Tempering ( REST2 ). J. Phys. Chem. 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [Green Version]
- Stirnemann, G.; Sterpone, F. Recovering Protein Thermal Stability Using All-Atom Hamiltonian Replica-Exchange Simulations in Explicit Solvent. J. Chem. Theory Comput. 2015, 11, 5573–5577. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.; Biswas, P. Estimating the mean first passage time of protein misfolding. Phys. Chem. Chem. Phys. 2018, 20, 5692–5698. [Google Scholar] [CrossRef]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Stirnemann, G.; Sterpone, F. Mechanics of Protein Adaptation to High Temperatures. J. Phys. Chem. Lett. 2017, 8, 5884–5890. [Google Scholar] [CrossRef] [PubMed]
- Katava, M.; Stirnemann, G.; Zanatta, M.; Capaccioli, S.; Pachetti, M.; Ngai, K.L.; Sterpone, F.; Paciaroni, A. Critical structural fluctuations of proteins upon thermal unfolding challenge the Lindemann criterion. Proc. Natl. Acad. Sci. USA 2017, 114, 9361–9366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timr, S.; Sterpone, F. Stabilizing or Destabilizing: Simulations of Chymotrypsin Inhibitor 2 under Crowding Reveal Existence of a Crossover Temperature. J. Phys. Chem. Lett. 2021, 12, 1741–1746. [Google Scholar] [CrossRef] [PubMed]
- Katava, M.; Stirnemann, G.; Pachetti, M.; Capaccioli, S.; Paciaroni, A.; Sterpone, F. Specific Interactions and Environment Flexibility Tune Protein Stability under Extreme Crowding. J. Phys. Chem. 2021, 125, 6103–6111. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald-an N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hockney, R.W.; Goel, S.P.; Eastwood, J.W. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys. 1974, 14, 148–158. [Google Scholar] [CrossRef]
- Gapsys, V.; De Groot, B.L.; Briones, R. Computational analysis of local membrane properties. J. -Comput.-Aided Mol. Des. 2013, 27, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeliger, D.; de Groot, B.L. Protein thermostability calculations using alchemical free energy simulations. Biophys. J. 2010, 98, 2309–2316. [Google Scholar] [CrossRef] [Green Version]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Prevost, M.; Wodak, S.J.; Tidor, B.; Karplus, M. Contribution of the hydrophobic effect to protein stability: Analysis based on simulations of the Ile-96 → Ala mutation in barnase. Proc. Natl. Acad. Sci. USA 1991, 88, 10880–10884. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, R.B.; Hummer, G. Optimized Molecular Dynamics Force Fields Applied to the Helix–Coil Transition of Polypeptides. J. Phys. Chem. 2009, 113, 9004–9015. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teilum, K.; Smith, M.H.; Schulz, E.; Christensen, L.C.; Solomentsev, G.; Oliveberg, M.; Akke, M. Transient structural distortion of metal-free Cu/Zn superoxide dismutase triggers aberrant oligomerization. Proc. Natl. Acad. Sci. USA 2009, 106, 18273–18278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, C.M.; Rumfeldt, J.A.; Broom, H.R.; Sekhar, A.; Kay, L.E.; Meiering, E.M. Concurrent Increases and Decreases in Local Stability and Conformational Heterogeneity in Cu, Zn Superoxide Dismutase Variants Revealed by Temperature-Dependence of Amide Chemical Shifts. Biochemistry 2016, 55, 1346–1361. [Google Scholar] [CrossRef]
- Kayatekin, C.; Cohen, N.R.; Matthews, C.R. Enthalpic barriers dominate the folding and unfolding of the human Cu, Zn superoxide dismutase monomer. J. Mol. Biol. 2012, 424, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Sen Mojumdar, S.; Scholl, Z.N.; Dee, D.R.; Rouleau, L.; Anand, U.; Garen, C.; Woodside, M.T. Partially native intermediates mediate misfolding of SOD1 in single-molecule folding trajectories. Nat. Commun. 2017, 8, 1881. [Google Scholar] [CrossRef]
- Ruff, K.M.; Choi, Y.H.; Cox, D.; Ormsby, A.R.; Myung, Y.; Ascher, D.B.; Radford, S.E.; Pappu, R.V.; Hatters, D.M. Sequence grammar underlying unfolding and phase separation of globular proteins. bioRxiv 2021. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timr, S.; Sterpone, F. Computational Insights into the Unfolding of a Destabilized Superoxide Dismutase 1 Mutant. Biology 2021, 10, 1240. https://doi.org/10.3390/biology10121240
Timr S, Sterpone F. Computational Insights into the Unfolding of a Destabilized Superoxide Dismutase 1 Mutant. Biology. 2021; 10(12):1240. https://doi.org/10.3390/biology10121240
Chicago/Turabian StyleTimr, Stepan, and Fabio Sterpone. 2021. "Computational Insights into the Unfolding of a Destabilized Superoxide Dismutase 1 Mutant" Biology 10, no. 12: 1240. https://doi.org/10.3390/biology10121240