
DRPPM-EASY: A Web-Based Framework for Integrative Analysis of Multi-Omics Cancer Datasets

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
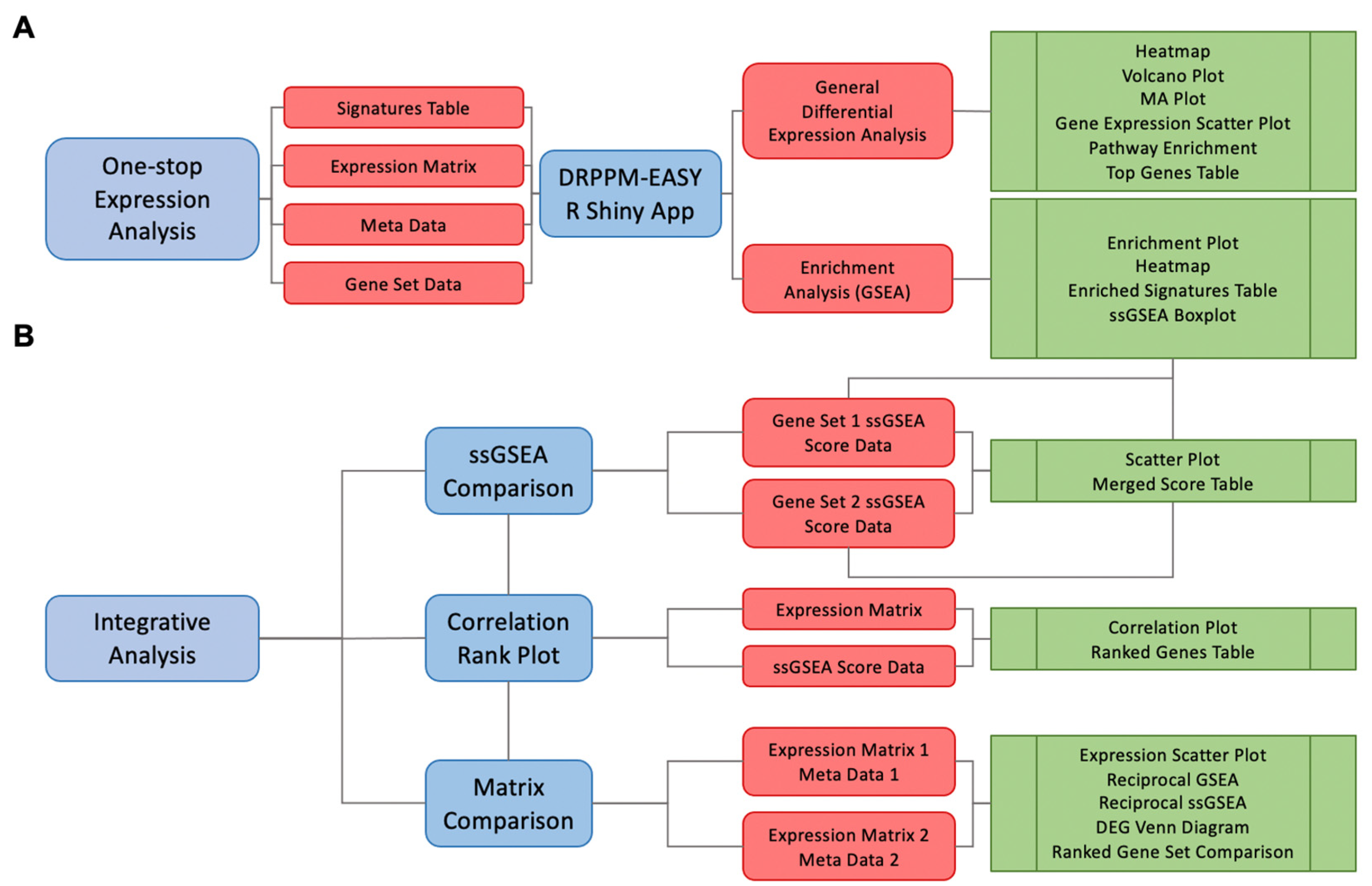
2.1. Module 1. DRPPM-EASY APP Implementation
2.2. Module 2. The DRPPM-EASY-Integration App Implementation
2.3. Installation and User Guide
2.4. RNA Sequencing Analysis
2.5. Whole Proteomics Mass Spectrometry and Data Analysis
2.6. Pre-Processing of the GSEA Analysis
3. Results
3.1. DRPPM-EASY Analysis of RNA-seq and Proteomics Data Use Case 1
3.2. DRPPM-EASY-CCLE Use Case 2
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Anda-Jauregui, G.; Hernandez-Lemus, E. Computational Oncology in the Multi-Omics Era: State of the Art. Front. Oncol. 2020, 10, 423. [Google Scholar] [CrossRef]
- Menyhart, O.; Gyorffy, B. Multi-omics approaches in cancer research with applications in tumor subtyping, prognosis, and diagnosis. Comput. Struct. Biotechnol. J. 2021, 19, 949–960. [Google Scholar] [CrossRef]
- Chai, A.W.Y.; Tan, A.C.; Cheong, S.C. Uncovering drug repurposing candidates for head and neck cancers: Insights from systematic pharmacogenomics data analysis. Sci. Rep. 2021, 11, 23933. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Wilks, C.; Zheng, S.C.; Chen, F.Y.; Charles, R.; Solomon, B.; Ling, J.P.; Imada, E.L.; Zhang, D.; Joseph, L.; Leek, J.T.; et al. Recount3: Summaries and queries for large-scale RNA-seq expression and splicing. Genome Biol. 2021, 22, 323. [Google Scholar] [CrossRef]
- Edwards, N.J.; Oberti, M.; Thangudu, R.R.; Cai, S.; McGarvey, P.B.; Jacob, S.; Madhavan, S.; Ketchum, K.A. The CPTAC Data Portal: A Resource for Cancer Proteomics Research. J. Proteome Res. 2015, 14, 2707–2713. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Davis-Turak, J.; Courtney, S.M.; Hazard, E.S.; Glen, W.B., Jr.; da Silveira, W.A.; Wesselman, T.; Harbin, L.P.; Wolf, B.J.; Chung, D.; Hardiman, G. Genomics pipelines and data integration: Challenges and opportunities in the research setting. Expert Rev. Mol. Diagn. 2017, 17, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Castellano-Escuder, P.; Gonzalez-Dominguez, R.; Carmona-Pontaque, F.; Andres-Lacueva, C.; Sanchez-Pla, A. POMAShiny: A user-friendly web-based workflow for metabolomics and proteomics data analysis. PLoS Comput. Biol. 2021, 17, e1009148. [Google Scholar] [CrossRef]
- Su, W.; Sun, J.; Shimizu, K.; Kadota, K. TCC-GUI: A Shiny-based application for differential expression analysis of RNA-Seq count data. BMC Res. Notes 2019, 12, 133. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Sklenar, J.; Barnes, A.P.; Minnier, J. The START App: A web-based RNAseq analysis and visualization resource. Bioinformatics 2017, 33, 447–449. [Google Scholar] [CrossRef]
- Reyes, A.L.P.; Silva, T.C.; Coetzee, S.G.; Plummer, J.T.; Davis, B.D.; Chen, S.; Hazelett, D.J.; Lawrenson, K.; Berman, B.P.; Gayther, S.A.; et al. GENAVi: A shiny web application for gene expression normalization, analysis and visualization. BMC Genom. 2019, 20, 745. [Google Scholar] [CrossRef]
- Zoppi, J.; Guillaume, J.F.; Neunlist, M.; Chaffron, S. MiBiOmics: An interactive web application for multi-omics data exploration and integration. BMC Bioinform. 2021, 22, 6. [Google Scholar] [CrossRef]
- Vanderwall, D.; Suresh, P.; Fu, Y.; Cho, J.H.; Shaw, T.I.; Mishra, A.; High, A.A.; Peng, J.; Li, Y. JUMPn: A Streamlined Application for Protein Co-Expression Clustering and Network Analysis in Proteomics. J. Vis. Exp. 2021, 176. [Google Scholar] [CrossRef]
- Dumas, J.; Gargano, M.A.; Dancik, G.M. shinyGEO: A web-based application for analyzing gene expression omnibus datasets. Bioinformatics 2016, 32, 3679–3681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toro-Dominguez, D.; Martorell-Marugan, J.; Lopez-Dominguez, R.; Garcia-Moreno, A.; Gonzalez-Rumayor, V.; Alarcon-Riquelme, M.E.; Carmona-Saez, P. ImaGEO: Integrative gene expression meta-analysis from GEO database. Bioinformatics 2019, 35, 880–882. [Google Scholar] [CrossRef]
- Shimada, K.; Bachman, J.A.; Muhlich, J.L.; Mitchison, T.J. ShinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. Elife 2021, 10, e57116. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, T.I.; Dong, L.; Tian, L.; Qian, C.; Liu, Y.; Ju, B.; High, A.; Kavdia, K.; Pagala, V.R.; Shaner, B.; et al. Integrative network analysis reveals USP7 haploinsufficiency inhibits E-protein activity in pediatric T-lineage acute lymphoblastic leukemia (T-ALL). Sci. Rep. 2021, 11, 5154. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Xu, P.; Duong, D.M.; Peng, J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res. 2009, 8, 3944–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagala, V.R.; High, A.A.; Wang, X.; Tan, H.; Kodali, K.; Mishra, A.; Kavdia, K.; Xu, Y.; Wu, Z.; Peng, J. Quantitative protein analysis by mass spectrometry. Methods Mol. Biol. 2015, 1278, 281–305. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Wu, Z.; Wang, H.; Tan, H.; Peng, J. JUMP: A tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteom. 2014, 13, 3663–3673. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.H.; Fountain, M.D., Jr.; Fon Tacer, K.; Xia, F.; Bi, W.; Kang, S.H.; Patel, A.; Rosenfeld, J.A.; Le Caignec, C.; Isidor, B.; et al. USP7 Acts as a Molecular Rheostat to Promote WASH-Dependent Endosomal Protein Recycling and Is Mutated in a Human Neurodevelopmental Disorder. Mol. Cell 2015, 59, 956–969. [Google Scholar] [CrossRef] [Green Version]
- Jin, Q.; Martinez, C.A.; Arcipowski, K.M.; Zhu, Y.; Gutierrez-Diaz, B.T.; Wang, K.K.; Johnson, M.R.; Volk, A.G.; Wang, F.; Wu, J.; et al. USP7 Cooperates with NOTCH1 to Drive the Oncogenic Transcriptional Program in T-Cell Leukemia. Clin. Cancer Res. 2019, 25, 222–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J. The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adderley, H.; Blackhall, F.H.; Lindsay, C.R. KRAS-mutant non-small cell lung cancer: Converging small molecules and immune checkpoint inhibition. EBioMedicine 2019, 41, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. Elife 2016, 5, e18413. [Google Scholar] [CrossRef]
- Grabocka, E.; Bar-Sagi, D. Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell 2016, 167, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Sundararajan, Z.; Knoll, R.; Hombach, P.; Becker, M.; Schultze, J.L.; Ulas, T. Shiny-Seq: Advanced guided transcriptome analysis. BMC Res. Notes 2019, 12, 432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krassowski, M.; Das, V.; Sahu, S.K.; Misra, B.B. State of the Field in Multi-Omics Research: From Computational Needs to Data Mining and Sharing. Front. Genet. 2020, 11, 610798. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| App Function | Description | |
|---|---|---|
| E1 | Unsupervised Heatmap |
|
| E2 | Scatter Plot |
|
| E3 | Custom Heatmap |
|
| E4 | Box Plot |
|
| App Function | Description | |
|---|---|---|
| DEA1 | Volcano Plot |
|
| DEA2 | MA Plot |
|
| DEA4 | Pathway Enrichment Analysis |
|
| App Function | Description | |
|---|---|---|
| GA1 | Enrichment Plot |
|
| GA2 | Gene Expression Heatmap |
|
| GA3 | GSEA Summary Table |
|
| GA4 | Generate Summary Table |
|
| GA5 | ssGSEA Boxplots |
|
| App Function | Description | |
|---|---|---|
| IA1 | Scatter Plot Comparison |
|
| IA2 | Correlation Rank Plot |
|
| IA3 | Matrix Comparison File Upload |
|
| IA4 | Log2FC Comparison Scatter Plot |
|
| IA5 | Reciprocal GSEA |
|
| IA6 | Reciprocal ssGSEA |
|
| IA7 | Venn Diagram |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obermayer, A.; Dong, L.; Hu, Q.; Golden, M.; Noble, J.D.; Rodriguez, P.; Robinson, T.J.; Teng, M.; Tan, A.-C.; Shaw, T.I. DRPPM-EASY: A Web-Based Framework for Integrative Analysis of Multi-Omics Cancer Datasets. Biology 2022, 11, 260. https://doi.org/10.3390/biology11020260
Obermayer A, Dong L, Hu Q, Golden M, Noble JD, Rodriguez P, Robinson TJ, Teng M, Tan A-C, Shaw TI. DRPPM-EASY: A Web-Based Framework for Integrative Analysis of Multi-Omics Cancer Datasets. Biology. 2022; 11(2):260. https://doi.org/10.3390/biology11020260
Chicago/Turabian StyleObermayer, Alyssa, Li Dong, Qianqian Hu, Michael Golden, Jerald D. Noble, Paulo Rodriguez, Timothy J. Robinson, Mingxiang Teng, Aik-Choon Tan, and Timothy I. Shaw. 2022. "DRPPM-EASY: A Web-Based Framework for Integrative Analysis of Multi-Omics Cancer Datasets" Biology 11, no. 2: 260. https://doi.org/10.3390/biology11020260