
Cardiac Calcifications: Phenotypes, Mechanisms, Clinical and Prognostic Implications

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
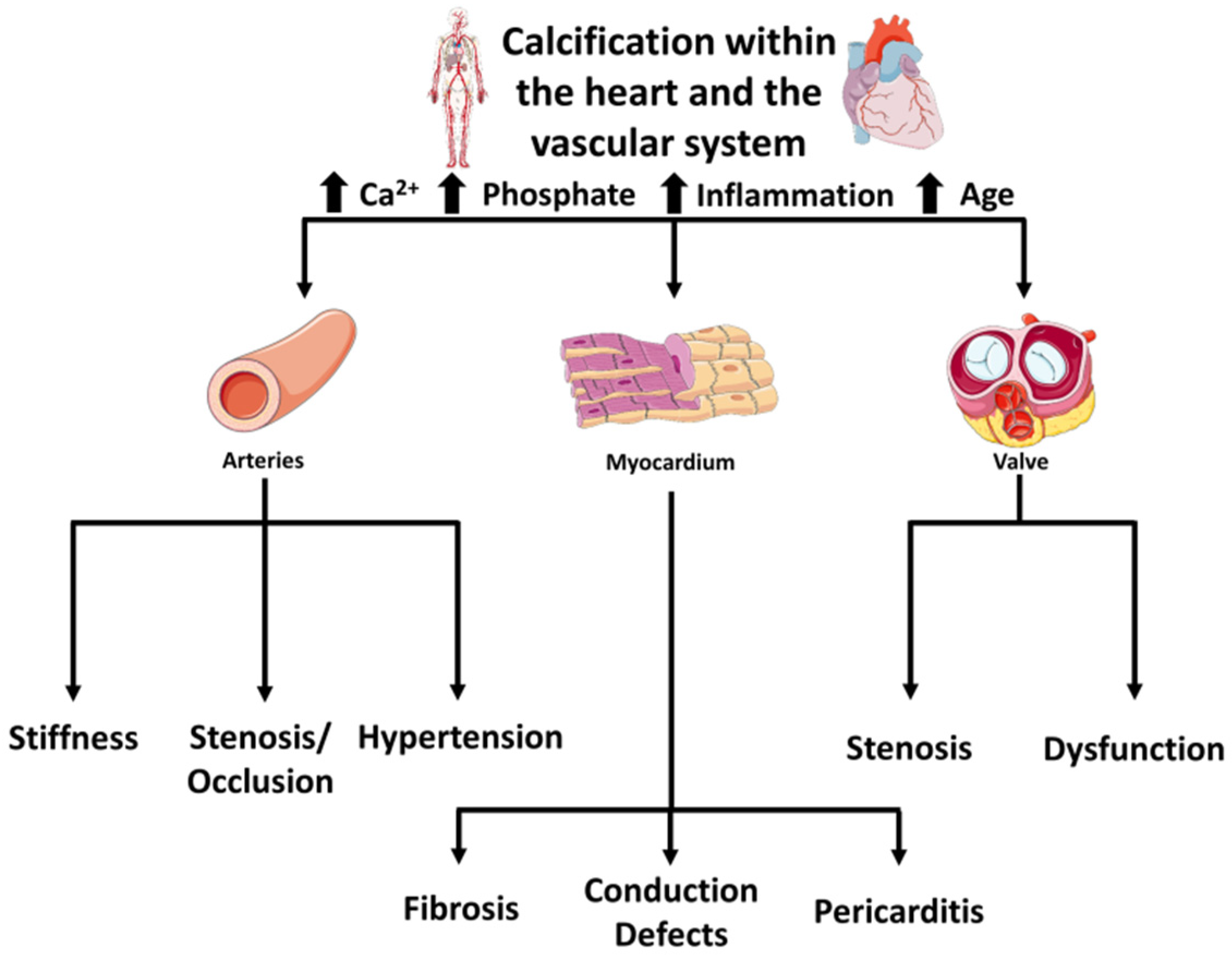
2. Diversity in Vascular Versus Valvular Calcification
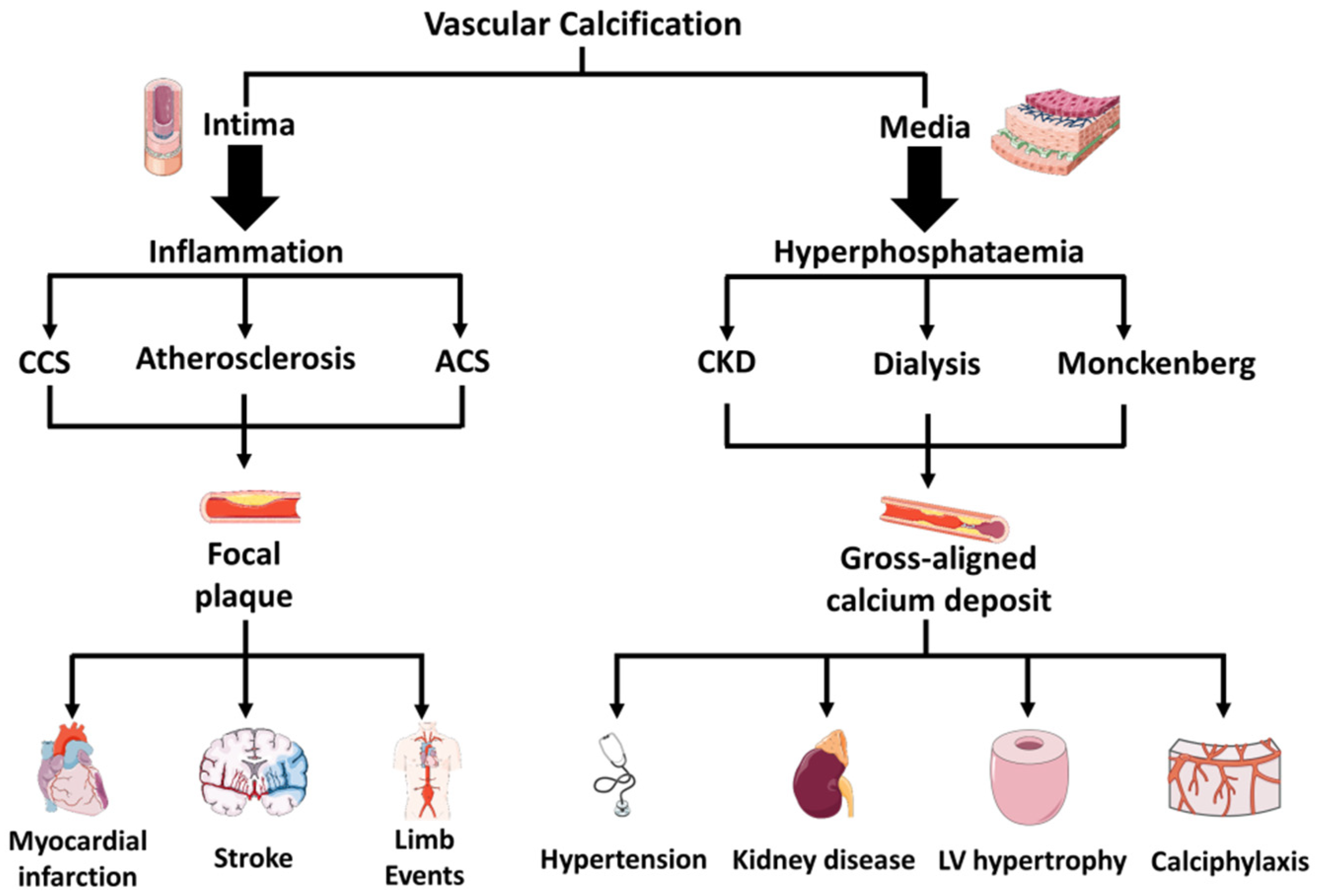
3. Diversity in Medial Versus Intimal Vascular Calcifications
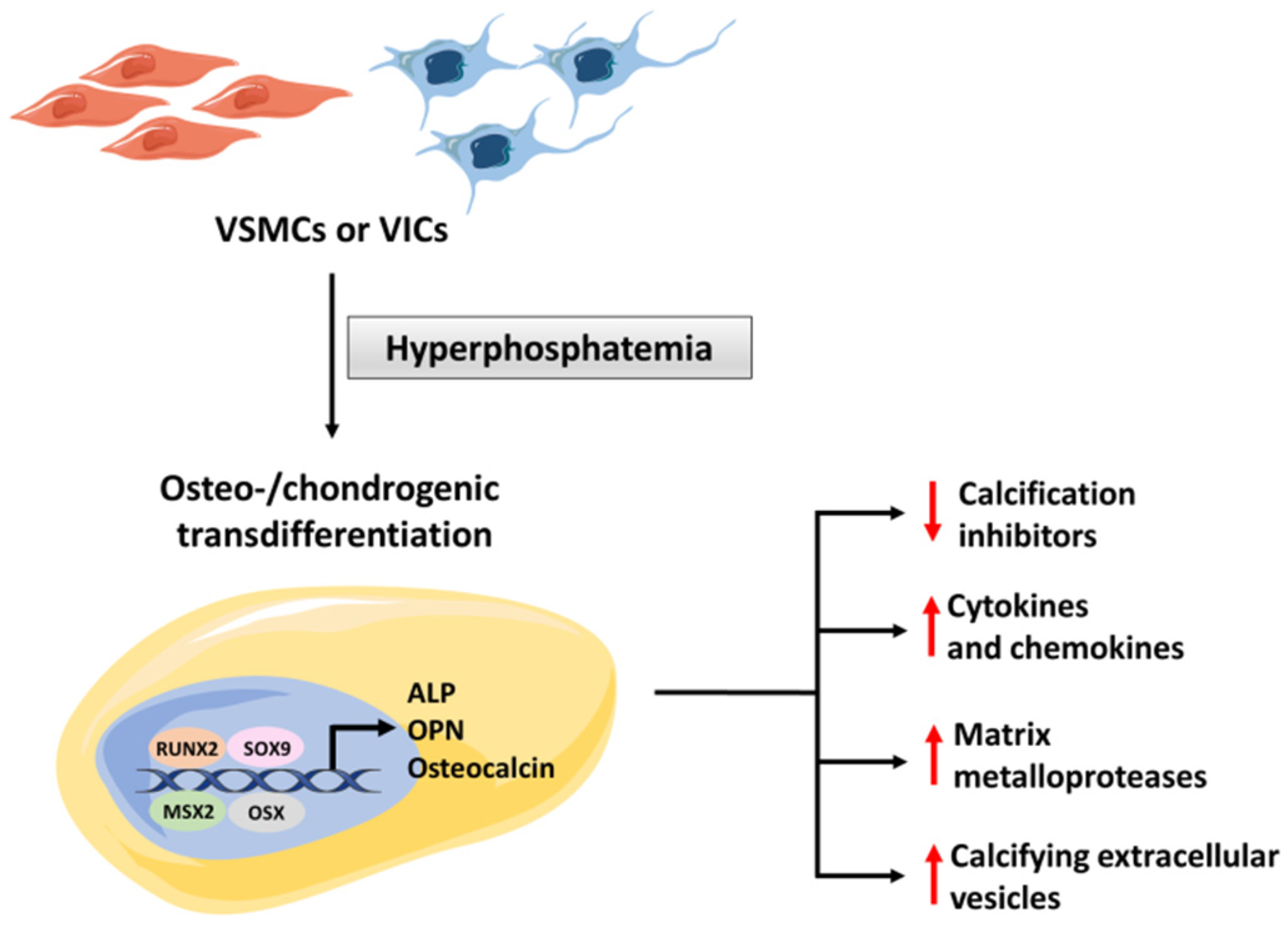
4. Mechanism of Hyperphosphatemic Medial Calcification
4.1. The Role of Osteogenic Transdifferentation
4.2. Regulatory Role of FGF-23 and Klotho
4.3. Regulatory Role of MGP, Fetuin-A and Calciprotein Particles
4.4. Regulatory Role of Extracellular Vesicles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Role | Actor | Description | Reference |
|---|---|---|---|
| Promoters of vascular calcification | TNAP | TNAP is an ectoenzyme that catalyzes dephosphorylations. Its activity releases free phosphate, which promotes mineralization. | [2] |
| RUNX2 | RUNX2 is a key transcription factor controlling osteoblast differentiation. | [5] | |
| OSX | OSX is a transcription factor necessary for osteocyte differentiation and bone formation | [5] | |
| NF-kB | NF-kB pathway plays an essential role in the transdifferentiation of VSMCs and VICs into osteochondrogenic cells. | [14,15] | |
| Osteocalcin | Osteocalcin is a hormone and osteogenic marker produced by osteoblast-like cells. | [16] | |
| SOX9 | SOX9 is a transcription factor associated with osteoblast-like transdifferentiation | [30] | |
| Secondary CPPs | Secondary CPPs are calcium hydroxyapatite nano-particles produced from primary CPPs under persistent hypercalcemia or hyperphosphatemia. | [36] | |
| Inhibitors of vascular calcification | FGF-23 | FGF-23 regulates phosphatemia by controlling renal phosphate excretion. Its role in vascular calcification is still debated. | [7] |
| Klotho | Klotho is a co-receptor essential for the binding of FGF-23 to its receptor. In addition, Klotho directly suppresses osteogenic transdifferentiation. | [30] | |
| MGP | MGP is a Gla-containing protein which binds calcium. It is secreted by VSMCs and acts as a potent inhibitor of vascular calcification. | [31] | |
| Fetuin-A | Fetuin-A is a glycoprotein secreted by the liver which binds calcium and phosphate, collaborating with MGP to prevent calcium precipitation in tissues. | [35] | |
| Primary CPPs | Primary CPPs are amorphous particles sized 50 to 500 nm that facilitate the clearance of calcium and phosphate, protecting from pathological calcification. | [36] | |
| CMVs | CMVs can promote or inhibit mineralization, depending on the phenotype of their originating cells and on the extracellular milieu. | [41] |
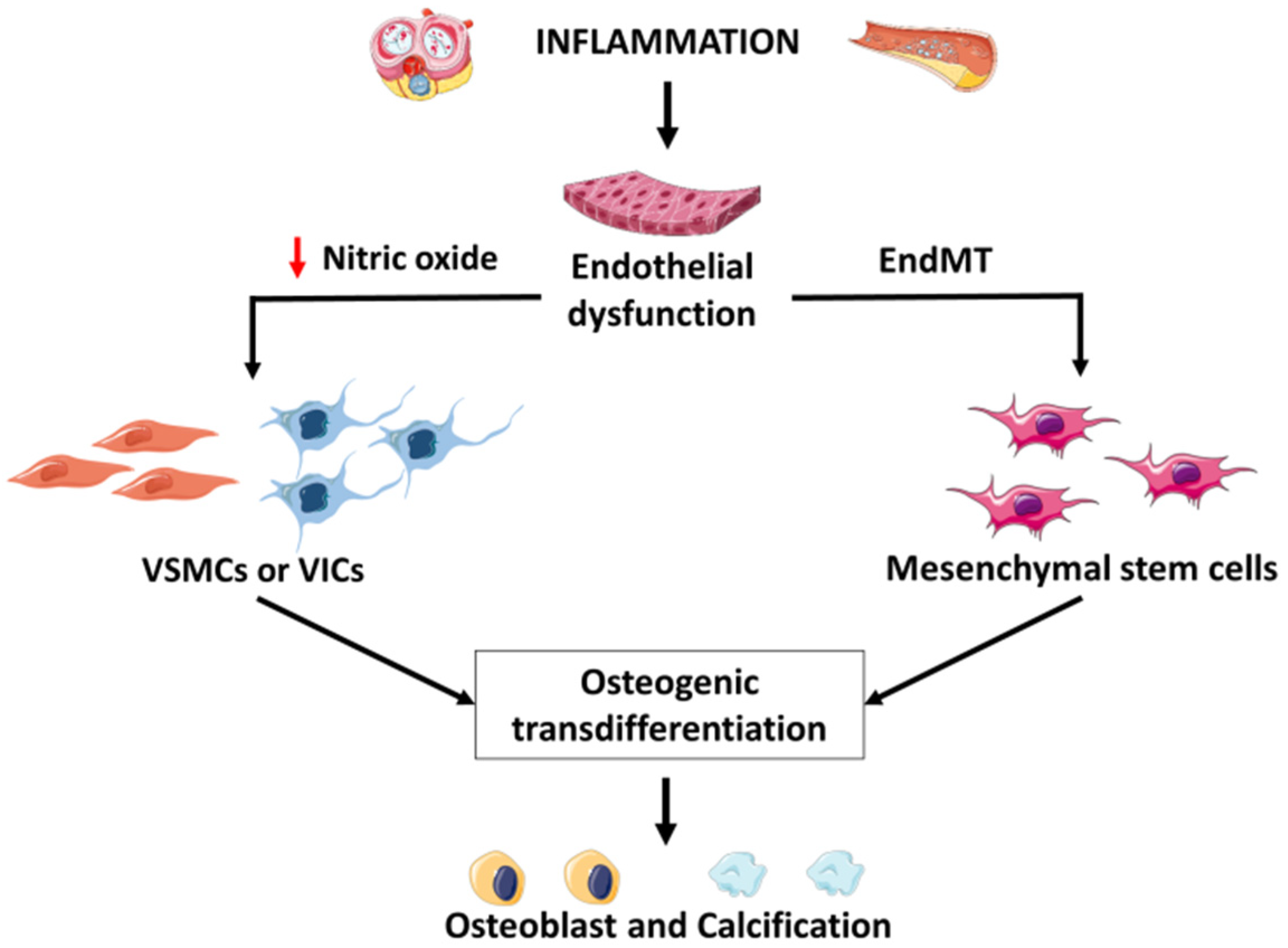
5. Mechanism of Inflammatory Intima Vascular Calcification
5.1. Role of Endothelial Cells
5.2. Regulatory Role of Shear Stress and of Notch and Wnt Signaling
5.3. Regulatory Role of Immune Cells
5.4. Regulatory Role of Clonal Hematopoiesis of Indeterminate Potential
6. Medial Calcification in Diabetes
7. Drug-Induced Vascular Calcification
7.1. Warfarin
7.2. Statins
7.3. COX-2 Inhibitors
8. Calcification Caused by Infections or Autoimmune Disorders
8.1. Infections
8.2. Autoimmune Diseases
9. Therapeutic Approaches
9.1. Treatment of Hyperphosphatemic Medial Vascular Calcification
9.2. Treatment of Inflammatory Intimal Vascular Calcification
10. Conclusions
11. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenland, P.; Blaha, M.J.; Budoff, M.J.; Erbel, R.; Watson, K.E. Coronary Calcium Score and Cardiovascular Risk. J. Am. Coll. Cardiol. 2018, 72, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Hutcheson, J.D.; Blaser, M.C.; Aikawa, E. Giving Calcification Its Due: Recognition of a Diverse Disease: A First Attempt to Standardize the Field. Circ. Res. 2017, 120, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, D.A.; Prasad, S.; Alotti, N. Calcific Aortic Valve Disease: Molecular Mechanisms and Therapeutic Approaches. Eur. Cardiol. 2015, 10, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieceli Dalla Sega, F.; Fortini, F.; Cimaglia, P.; Marracino, L.; Tonet, E.; Antonucci, A.; Moscarelli, M.; Campo, G.; Rizzo, P.; Ferrari, R. COX-2 Is Downregulated in Human Stenotic Aortic Valves and Its Inhibition Promotes Dystrophic Calcification. Int. J. Mol. Sci. 2020, 21, 8917. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Lang, F.; Eckardt, K.U.; Amann, K.; Kuro-O, M.; Pasch, A.; Pieske, B.; Alesutan, I. Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia. Cell. Mol. Life Sci. 2019, 76, 2077–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, C.H.; Saikrishnan, N.; Tamilselvan, G.; Vasilyev, N.; Yoganathan, A.P. The congenital bicuspid aortic valve can experience high-frequency unsteady shear stresses on its leaflet surface. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H721–H731. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.J.; Raggi, P.; Wolf, M.; Gold, A.M.; Chertow, G.M.; Roe, M.T. Targeting Vascular Calcification in Chronic Kidney Disease. JACC Basic Transl. Sci. 2020, 5, 398–412. [Google Scholar] [CrossRef]
- St. Hilaire, C. Medial Arterial Calcification: A Significant and Independent Contributor of Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 253–260. [Google Scholar] [CrossRef]
- García-Lozano, J.A.; Ocampo-Candiani, J.; Martínez-Cabriales, S.A.; Garza-Rodríguez, V. An Update on Calciphylaxis. Am. J. Clin. Dermatol. 2018, 19, 599–608. [Google Scholar] [CrossRef]
- Shu, J.; Santulli, G. Update on peripheral artery disease: Epidemiology and evidence-based facts. Atherosclerosis 2018, 275, 379–381. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Goettsch, C.; Bertazzo, S.; Maldonado, N.; Ruiz, J.L.; Goh, W.; Yabusaki, K.; Faits, T.; Bouten, C.; Franck, G.; et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016, 15, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergwitz, C.; Jüppner, H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 2010, 61, 91–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koumakis, E.; Cormier, C.; Roux, C.; Briot, K. The Causes of Hypo- and Hyperphosphatemia in Humans. Calcif. Tissue Int. 2021, 108, 41–73. [Google Scholar] [CrossRef]
- Yoshida, T.; Yamashita, M.; Horimai, C.; Hayashi, M. Smooth Muscle-Selective Nuclear Factor-κB Inhibition Reduces Phosphate-Induced Arterial Medial Calcification in Mice with Chronic Kidney Disease. J. Am. Heart Assoc. 2017, 6, e007248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, T.; Farrar, E.; Wang, Y.; Wu, B.; Hsu, K.; Zhou, B.; Butcher, J. NFκB (Nuclear Factor κ-Light-Chain Enhancer of Activated B Cells) Activity Regulates Cell-Type-Specific and Context-Specific Susceptibility to Calcification in the Aortic Valve. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 638–655. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- St. Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef]
- Jin, H.; St. Hilaire, C.; Huang, Y.; Yang, D.; Dmitrieva, N.I.; Negro, A.; Schwartzbeck, R.; Liu, Y.; Yu, Z.; Walts, A.; et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci. Signal. 2016, 9, ra121. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Tamada, T.; Kasai, N.; Urakawa, I.; Aono, Y.; Hasegawa, H.; Fujita, T.; Kuroki, R.; Yamashita, T.; Fukumoto, S.; et al. Anti-FGF23 neutralizing antibodies show the physiological role and structural features of FGF23. J. Bone Miner. Res. 2008, 23, 1509–1518. [Google Scholar] [CrossRef]
- Komaba, H.; Fuller, D.S.; Taniguchi, M.; Yamamoto, S.; Nomura, T.; Zhao, J.; Bieber, B.A.; Robinson, B.M.; Pisoni, R.L.; Fukagawa, M. Fibroblast Growth Factor 23 and Mortality Among Prevalent Hemodialysis Patients in the Japan Dialysis Outcomes and Practice Patterns Study. Kidney Int. Rep. 2020, 5, 1956–1964. [Google Scholar] [CrossRef]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Lu, T.-S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.-L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, D.; Mackenzie, N.C.; Millan, J.L.; Farquharson, C.; MacRae, V.E. A protective role for FGF-23 in local defence against disrupted arterial wall integrity? Mol. Cell. Endocrinol. 2013, 372, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimbo, R.; Kawakami-Mori, F.; Mu, S.; Hirohama, D.; Majtan, B.; Shimizu, Y.; Yatomi, Y.; Fukumoto, S.; Fujita, T.; Shimosawa, T. Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of Klotho deficiency. Kidney Int. 2014, 85, 1103–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scialla, J.J.; Xie, H.; Rahman, M.; Anderson, A.H.; Isakova, T.; Ojo, A.; Zhang, X.; Nessel, L.; Hamano, T.; Grunwald, J.E.; et al. Fibroblast growth factor-23 and cardiovascular events in CKD. J. Am. Soc. Nephrol. 2014, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.-C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef]
- Ohnishi, M.; Nakatani, T.; Lanske, B.; Razzaque, M.S. In vivo genetic evidence for suppressing vascular and soft-tissue calcification through the reduction of serum phosphate levels, even in the presence of high serum calcium and 1,25-dihydroxyvitamin d levels. Circ. Cardiovasc. Genet. 2009, 2, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xue, D.; Hu, D.; Xie, T.; Tao, Y.; Zhu, T.; Chen, E.; Pan, Z. Secreted klotho protein attenuates osteogenic differentiation of human bone marrow mesenchymal stem cells in vitro via inactivation of the FGFR1/ERK signaling pathway. Growth Factors 2015, 33, 356–365. [Google Scholar] [CrossRef]
- Li, F.; Yao, Q.; Ao, L.; Cleveland, J.C.; Dong, N.; Fullerton, D.A.; Meng, X. Klotho suppresses high phosphate-induced osteogenic responses in human aortic valve interstitial cells through inhibition of Sox9. J. Mol. Med. 2017, 95, 739–751. [Google Scholar] [CrossRef]
- Munroe, P.B.; Olgunturk, R.O.; Fryns, J.P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.; Gardiner, R.M.; Chung, E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Wajih, N.; Borras, T.; Xue, W.; Hutson, S.M.; Wallin, R. Processing and transport of matrix gamma-carboxyglutamic acid protein and bone morphogenetic protein-2 in cultured human vascular smooth muscle cells: Evidence for an uptake mechanism for serum fetuin. J. Biol. Chem. 2004, 279, 43052–43060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, M.; Babler, A.; Moshkova, I.; Gremse, F.; Kiessling, F.; Kusebauch, U.; Nelea, V.; Kramann, R.; Moritz, R.L.; McKee, M.D.; et al. Lumenal calcification and microvasculopathy in fetuin-A-deficient mice lead to multiple organ morbidity. PLoS ONE 2020, 15, e0228503. [Google Scholar] [CrossRef] [Green Version]
- Westenfeld, R.; Schäfer, C.; Krüger, T.; Haarmann, C.; Schurgers, L.J.; Reutelingsperger, C.; Ivanovski, O.; Drueke, T.; Massy, Z.A.; Ketteler, M.; et al. Fetuin-A protects against atherosclerotic calcification in CKD. J. Am. Soc. Nephrol. 2009, 20, 1264–1274. [Google Scholar] [CrossRef] [Green Version]
- Kutikhin, A.G.; Feenstra, L.; Kostyunin, A.E.; Yuzhalin, A.E.; Hillebrands, J.L.; Krenning, G. Calciprotein Particles: Balancing Mineral Homeostasis and Vascular Pathology. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1607–1624. [Google Scholar] [CrossRef]
- Aikawa, E.; Blaser, M.C. 2020 Jeffrey M. Hoeg Award Lecture: Calcifying Extracellular Vesicles as Building Blocks of Microcalcifications in Cardiovascular Disorders. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 117–127. [Google Scholar] [CrossRef]
- Kutikhin, A.G.; Velikanova, E.A.; Mukhamadiyarov, R.A.; Glushkova, T.V.; Borisov, V.V.; Matveeva, V.G.; Antonova, L.V.; Filip’ev, D.E.; Golovkin, A.S.; Shishkova, D.K.; et al. Apoptosis-mediated endothelial toxicity but not direct calcification or functional changes in anti-calcification proteins defines pathogenic effects of calcium phosphate bions. Sci. Rep. 2016, 6, 27255. [Google Scholar] [CrossRef]
- Shishkova, D.; Markova, V.; Sinitsky, M.; Tsepokina, A.; Velikanova, E.; Bogdanov, L.; Glushkova, T.; Kutikhin, A. Calciprotein Particles Cause Endothelial Dysfunction under Flow. Int. J. Mol. Sci. 2020, 21, 8802. [Google Scholar] [CrossRef]
- Patidar, A.; Singh, D.K.; Winocour, P.; Farrington, K.; Baydoun, A.R. Human uraemic serum displays calcific potential in vitro that increases with advancing chronic kidney disease. Clin. Sci. 2013, 125, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lee, I.-K.; Jeon, J.-H. Vascular Calcification-New Insights into Its Mechanism. Int. J. Mol. Sci. 2020, 21, 2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, S.C.; Khalid, S.; Smethurst, V.; Monier, D.; Mobley, J.; Huet, A.; Conway, J.F.; Napierala, D. Proteomic profiling of extracellular vesicles released from vascular smooth muscle cells during initiation of phosphate-induced mineralization. Connect. Tissue Res. 2018, 59, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, M.A.; Buffolo, F.; Schlotter, F.; Atkins, S.K.; Lee, L.H.; Halu, A.; Blaser, M.C.; Tsolaki, E.; Higashi, H.; Luther, K.; et al. Annexin A1-dependent tethering promotes extracellular vesicle aggregation revealed with single-extracellular vesicle analysis. Sci. Adv. 2020, 6, eabb1244. [Google Scholar] [CrossRef]
- Kanno, Y.; Into, T.; Lowenstein, C.J.; Matsushita, K. Nitric oxide regulates vascular calcification by interfering with TGF-β signalling. Cardiovasc. Res. 2008, 77, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-C.; Yen, M.-H.; Yen, C.-H.; Lau, Y.-T. Oxidized low density lipoprotein induces apoptosis via generation of reactive oxygen species in vascular smooth muscle cells. Cardiovasc. Res. 2001, 49, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.; Lee, S.; Kim, S.-M.; Lee, E.-J.; Lee, S.-R.; Kim, D.-H.; Jang, J.-Y.; Kang, S.-W.; Lee, K.-U.; Chang, E.-J.; et al. Dipeptidyl Peptidase-4 Induces Aortic Valve Calcification by Inhibiting Insulin-Like Growth Factor-1 Signaling in Valvular Interstitial Cells. Circulation 2017, 135, 1935–1950. [Google Scholar] [CrossRef]
- Hortells, L.; Sur, S.; St. Hilaire, C. Cell Phenotype Transitions in Cardiovascular Calcification. Front. Cardiovasc. Med. 2018, 5, 27. [Google Scholar] [CrossRef]
- Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Guo, Y.; Moon, J.H.; Jumabay, M.; Boström, K.I.; Yao, Y. Serine Protease Activation Essential for Endothelial-Mesenchymal Transition in Vascular Calcification. Circ. Res. 2015, 117, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Fernandez Esmerats, J.; Villa-Roel, N.; Kumar, S.; Gu, L.; Salim, M.T.; Ohh, M.; Taylor, W.R.; Nerem, R.M.; Yoganathan, A.P.; Jo, H. Disturbed Flow Increases UBE2C (Ubiquitin E2 Ligase C) via Loss of miR-483-3p, Inducing Aortic Valve Calcification by the pVHL (von Hippel-Lindau Protein) and HIF-1α (Hypoxia-Inducible Factor-1α) Pathway in Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 467–481. [Google Scholar] [CrossRef] [Green Version]
- Souilhol, C.; Serbanovic-Canic, J.; Fragiadaki, M.; Chico, T.J.; Ridger, V.; Roddie, H.; Evans, P.C. Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 2020, 17, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Pektas, M.B.; Koca, H.B.; Sadi, G.; Akar, F. Dietary Fructose Activates Insulin Signaling and Inflammation in Adipose Tissue: Modulatory Role of Resveratrol. Biomed Res. Int. 2016, 2016, 8014252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoris, C.V.; Li, M.; White, M.P.; Liu, L.; He, D.; Pollard, K.S.; Bruneau, B.G.; Srivastava, D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 2015, 160, 1072–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.P.; Theodoris, C.V.; Liu, L.; Collins, W.J.; Blue, K.W.; Lee, J.H.; Meng, X.; Robbins, R.C.; Ivey, K.N.; Srivastava, D. NOTCH1 regulates matrix gla protein and calcification gene networks in human valve endothelium. J. Mol. Cell. Cardiol. 2015, 84, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Albanese, I.; Khan, K.; Barratt, B.; Al-Kindi, H.; Schwertani, A. Atherosclerotic Calcification: Wnt Is the Hint. J. Am. Heart Assoc. 2018, 7, e007356. [Google Scholar] [CrossRef] [Green Version]
- Aquila, G.; Kostina, A.; Vieceli Dalla Sega, F.; Shlyakhto, E.; Kostareva, A.; Marracino, L.; Ferrari, R.; Rizzo, P.; Malaschicheva, A. The Notch pathway: A novel therapeutic target for cardiovascular diseases? Expert Opin. Ther. Targets 2019, 23, 695–710. [Google Scholar] [CrossRef]
- Bosse, K.; Hans, C.P.; Zhao, N.; Koenig, S.N.; Huang, N.; Guggilam, A.; LaHaye, S.; Tao, G.; Lucchesi, P.A.; Lincoln, J.; et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J. Mol. Cell. Cardiol. 2013, 60, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, U.; Manivannan, S.; Basu, M.; Ueyama, Y.; Blaser, M.C.; Cameron, E.; McDermott, M.R.; Lincoln, J.; Cole, S.E.; Wood, S.; et al. Nitric oxide prevents aortic valve calcification by S-nitrosylation of USP9X to activate NOTCH signaling. Sci. Adv. 2021, 7, eabe3706. [Google Scholar] [CrossRef]
- Kostina, A.; Shishkova, A.; Ignatieva, E.; Irtyuga, O.; Bogdanova, M.; Levchuk, K.; Golovkin, A.; Zhiduleva, E.; Uspenskiy, V.; Moiseeva, O.; et al. Different Notch signaling in cells from calcified bicuspid and tricuspid aortic valves. J. Mol. Cell. Cardiol. 2018, 114, 211–219. [Google Scholar] [CrossRef]
- Shimizu, T.; Tanaka, T.; Iso, T.; Matsui, H.; Ooyama, Y.; Kawai-Kowase, K.; Arai, M.; Kurabayashi, M. Notch signaling pathway enhances bone morphogenetic protein 2 (BMP2) responsiveness of Msx2 gene to induce osteogenic differentiation and mineralization of vascular smooth muscle cells. J. Biol. Chem. 2011, 286, 19138–19148. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liu, X.; Zhang, Y.; Di, M.; Wang, H.; Wang, L.; Chen, Y.; Cao, X.; Zeng, R.; Zhang, M. Upregulation of Dickkopf1 by oscillatory shear stress accelerates atherogenesis. J. Mol. Med. 2016, 94, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-L.; Shao, J.-S.; Behrmann, A.; Krchma, K.; Towler, D.A. Dkk1 and MSX2-Wnt7b signaling reciprocally regulate the endothelial-mesenchymal transition in aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1679–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafer, S.L.; Towler, D.A. Transcriptional regulation of SM22alpha by Wnt3a: Convergence with TGFbeta(1)/Smad signaling at a novel regulatory element. J. Mol. Cell. Cardiol. 2009, 46, 621–635. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-L.; Shao, J.-S.; Cai, J.; Sierra, O.L.; Towler, D.A. Msx2 exerts bone anabolism via canonical Wnt signaling. J. Biol. Chem. 2008, 283, 20505–20522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boström, K.I.; Rajamannan, N.M.; Towler, D.A. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ. Res. 2011, 109, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.-S.; Cheng, S.-L.; Pingsterhaus, J.M.; Charlton-Kachigian, N.; Loewy, A.P.; Towler, D.A. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J. Clin. Investig. 2005, 115, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Passos, L.S.A.; Lupieri, A.; Becker-Greene, D.; Aikawa, E. Innate and adaptive immunity in cardiovascular calcification. Atherosclerosis 2020, 306, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Vieceli Dalla Sega, F.; Fortini, F.; Aquila, G.; Campo, G.; Vaccarezza, M.; Rizzo, P. Notch Signaling Regulates Immune Responses in Atherosclerosis. Front. Immunol. 2019, 10, 1130. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zhang, Y.; Feng, W.; Chen, R.; Chen, J.; Touyz, R.M.; Wang, J.; Huang, H. Interleukin-18 Enhances Vascular Calcification and Osteogenic Differentiation of Vascular Smooth Muscle Cells Through TRPM7 Activation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Hecht, E.; Freise, C.; Websky, K.V.; Nasser, H.; Kretzschmar, N.; Stawowy, P.; Hocher, B.; Querfeld, U. The matrix metalloproteinases 2 and 9 initiate uraemic vascular calcifications. Nephrol. Dial. Transpl. 2016, 31, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Simionescu, A.; Simionescu, D.T.; Vyavahare, N.R. Osteogenic responses in fibroblasts activated by elastin degradation products and transforming growth factor-beta1: Role of myofibroblasts in vascular calcification. Am. J. Pathol. 2007, 171, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Schrader, J.; Niebergall, U.; Viereck, V.; Burchert, A.; Hörsch, D.; Preissner, K.T.; Schoppet, M. Interleukin-4 differentially regulates osteoprotegerin expression and induces calcification in vascular smooth muscle cells. Thromb. Haemost. 2006, 95, 708–714. [Google Scholar] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Papa, V.; Marracino, L.; Fortini, F.; Rizzo, P.; Campo, G.; Vaccarezza, M.; Vieceli Dalla Sega, F. Translating Evidence from Clonal Hematopoiesis to Cardiovascular Disease: A Systematic Review. J. Clin. Med. 2020, 9, 2480. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Mas-Peiro, S.; Hoffmann, J.; Fichtlscherer, S.; Dorsheimer, L.; Rieger, M.A.; Dimmeler, S.; Vasa-Nicotera, M.; Zeiher, A.M. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur. Heart J. 2020, 41, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Allison, M.A.; Hsi, S.; Wassel, C.L.; Morgan, C.; Ix, J.H.; Wright, C.M.; Criqui, M.H. Calcified atherosclerosis in different vascular beds and the risk of mortality. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 140–146. [Google Scholar] [CrossRef] [Green Version]
- McClelland, R.L.; Chung, H.; Detrano, R.; Post, W.; Kronmal, R.A. Distribution of coronary artery calcium by race, gender, and age: Results from the Multi-Ethnic Study of Atherosclerosis (MESA). Circulation 2006, 113, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Sun, Z.; Zang, G.; Zhang, L.; Hou, L.; Shao, C.; Wang, Z. Epidemiological Research Advances in Vascular Calcification in Diabetes. J. Diabetes Res. 2021, 2021, 4461311. [Google Scholar] [CrossRef]
- Kakani, E.; Elyamny, M.; Ayach, T.; El-Husseini, A. Pathogenesis and management of vascular calcification in CKD and dialysis patients. Semin. Dial. 2019, 32, 553–561. [Google Scholar] [CrossRef]
- Düsing, P.; Zietzer, A.; Goody, P.R.; Hosen, M.R.; Kurts, C.; Nickenig, G.; Jansen, F. Vascular pathologies in chronic kidney disease: Pathophysiological mechanisms and novel therapeutic approaches. J. Mol. Med. 2021, 99, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Kennon, A.M.; Stewart, J.A. RAGE Differentially Altered. Front. Physiol. 2021, 12, 676727. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Luo, D.; He, W.; Chen, J.; Su, X.; Huang, H. Diabetes and calcification: The potential role of anti-diabetic drugs on vascular calcification regression. Pharmacol. Res. 2020, 158, 104861. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef]
- Poetsch, F.; Henze, L.A.; Estepa, M.; Moser, B.; Pieske, B.; Lang, F.; Eckardt, K.U.; Alesutan, I.; Voelkl, J. Role of SGK1 in the Osteogenic Transdifferentiation and Calcification of Vascular Smooth Muscle Cells Promoted by Hyperglycemic Conditions. Int. J. Mol. Sci. 2020, 21, 7207. [Google Scholar] [CrossRef]
- Heath, J.M.; Sun, Y.; Yuan, K.; Bradley, W.E.; Litovsky, S.; Dell’Italia, L.J.; Chatham, J.C.; Wu, H.; Chen, Y. Activation of AKT by O-linked N-acetylglucosamine induces vascular calcification in diabetes mellitus. Circ. Res. 2014, 114, 1094–1102. [Google Scholar] [CrossRef] [Green Version]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Schiro, A.; Inglott, F.S.; Alexander, M.Y.; Weston, R. Suppression of SIRT1 in Diabetic Conditions Induces Osteogenic Differentiation of Human Vascular Smooth Muscle Cells via RUNX2 Signalling. Sci. Rep. 2019, 9, 878. [Google Scholar] [CrossRef]
- Bartoli-Leonard, F.; Wilkinson, F.L.; Schiro, A.; Serracino Inglott, F.; Alexander, M.Y.; Weston, R. Loss of SIRT1 in diabetes accelerates DNA damage-induced vascular calcification. Cardiovasc. Res. 2021, 117, 836–849. [Google Scholar] [CrossRef]
- Andrews, J.; Psaltis, P.J.; Bayturan, O.; Shao, M.; Stegman, B.; Elshazly, M.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E.; Nicholls, S.J.; et al. Warfarin Use Is Associated with Progressive Coronary Arterial Calcification: Insights from Serial Intravascular Ultrasound. JACC Cardiovasc. Imaging 2018, 11, 1315–1323. [Google Scholar] [CrossRef]
- Nuotio, K.; Koskinen, S.M.; Mäkitie, L.; Tuimala, J.; Ijäs, P.; Heikkilä, H.M.; Saksi, J.; Vikatmaa, P.; Sorto, P.; Kasari, S.; et al. Warfarin Treatment Is Associated to Increased Internal Carotid Artery Calcification. Front. Neurol. 2021, 12, 696244. [Google Scholar] [CrossRef]
- Han, K.H.; O’Neill, W.C. Increased Peripheral Arterial Calcification in Patients Receiving Warfarin. J. Am. Heart Assoc. 2016, 5, e002665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elango, K.; Javaid, A.; Khetarpal, B.K.; Ramalingam, S.; Kolandaivel, K.P.; Gunasekaran, K.; Ahsan, C. The Effects of Warfarin and Direct Oral Anticoagulants on Systemic Vascular Calcification: A Review. Cells 2021, 10, 773. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Cranenburg, E.C.; Vermeer, C. Matrix Gla-protein: The calcification inhibitor in need of vitamin K. Thromb. Haemost. 2008, 100, 593–603. [Google Scholar] [PubMed]
- Qiu, M.; Lu, Y.; Li, J.; Gu, J.; Ji, Y.; Shao, Y.; Kong, X.; Sun, W. Interaction of SOX5 with SOX9 promotes warfarin-induced aortic valve interstitial cell calcification by repressing transcriptional activation of LRP6. J. Mol. Cell. Cardiol. 2022, 162, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Henein, M.; Granåsen, G.; Wiklund, U.; Schmermund, A.; Guerci, A.; Erbel, R.; Raggi, P. High dose and long-term statin therapy accelerate coronary artery calcification. Int. J. Cardiol. 2015, 184, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Nicholls, S.J.; Shao, M.; Kataoka, Y.; Uno, K.; Kapadia, S.R.; Tuzcu, E.M.; Nissen, S.E. Impact of statins on serial coronary calcification during atheroma progression and regression. J. Am. Coll. Cardiol. 2015, 65, 1273–1282. [Google Scholar] [CrossRef]
- Xian, J.Z.; Lu, M.; Fong, F.; Qiao, R.; Patel, N.R.; Abeydeera, D.; Iriana, S.; Demer, L.L.; Tintut, Y. Statin Effects on Vascular Calcification: Microarchitectural Changes in Aortic Calcium Deposits in Aged Hyperlipidemic Mice. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e185–e192. [Google Scholar] [CrossRef] [PubMed]
- Bowler, M.A.; Raddatz, M.A.; Johnson, C.L.; Lindman, B.R.; Merryman, W.D. Celecoxib Is Associated with Dystrophic Calcification and Aortic Valve Stenosis. JACC Basic Transl. Sci. 2019, 4, 135–143. [Google Scholar] [CrossRef]
- Davutoglu, V.; Celik, A.; Aksoy, M. Contribution of selected serum inflammatory mediators to the progression of chronic rheumatic valve disease, subsequent valve calcification and NYHA functional class. J. Heart Valve Dis. 2005, 14, 251–256. [Google Scholar]
- Rajamannan, N.M.; Nealis, T.B.; Subramaniam, M.; Pandya, S.; Stock, S.R.; Ignatiev, C.I.; Sebo, T.J.; Rosengart, T.K.; Edwards, W.D.; McCarthy, P.M.; et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation 2005, 111, 3296–3301. [Google Scholar] [CrossRef] [Green Version]
- Cohen, D.J.; Malave, D.; Ghidoni, J.J.; Iakovidis, P.; Everett, M.M.; You, S.; Liu, Y.; Boyan, B.D. Role of oral bacterial flora in calcific aortic stenosis: An animal model. Ann. Thorac. Surg. 2004, 77, 537–543. [Google Scholar] [CrossRef]
- Beukers, N.G.; van der Heijden, G.J.; van Wijk, A.J.; Loos, B.G. Periodontitis is an independent risk indicator for atherosclerotic cardiovascular diseases among 60 174 participants in a large dental school in the Netherlands. J. Epidemiol. Community Health 2017, 71, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.J.; Choi, S.H.; Kim, D.; Kang, S.J.; Chung, S.J.; Choi, S.Y.; Yoon, D.H.; Lim, S.H.; Kim, Y.S.; Yim, J.Y.; et al. Association between Helicobacter pylori Seropositivity and the Coronary Artery Calcium Score in a Screening Population. Gut Liver 2011, 5, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Katz, R.J.; Quyyumi, A.A.; Canos, D.A.; Rott, D.; Csako, G.; Zalles-Ganley, A.; Ogunmakinwa, J.; Wasserman, A.G.; Epstein, S.E. Association of serum antibodies to heat-shock protein 65 with coronary calcification levels: Suggestion of pathogen-triggered autoimmunity in early atherosclerosis. Circulation 2004, 109, 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frostegård, J. Atherosclerosis in patients with autoimmune disorders. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1776–1785. [Google Scholar] [CrossRef]
- Paccou, J.; Brazier, M.; Mentaverri, R.; Kamel, S.; Fardellone, P.; Massy, Z.A. Vascular calcification in rheumatoid arthritis: Prevalence, pathophysiological aspects and potential targets. Atherosclerosis 2012, 224, 283–290. [Google Scholar] [CrossRef]
- Geraldino-Pardilla, L.; Giles, J.T.; Sokolove, J.; Zartoshti, A.; Robinson, W.H.; Budoff, M.; Detrano, R.; Bokhari, S.; Bathon, J.M. Association of Anti-Citrullinated Peptide Antibodies with Coronary Artery Calcification in Rheumatoid Arthritis. Arthritis Care Res. 2017, 69, 1276–1281. [Google Scholar] [CrossRef]
- Asanuma, Y.; Chung, C.P.; Oeser, A.; Solus, J.F.; Avalos, I.; Gebretsadik, T.; Shintani, A.; Raggi, P.; Sokka, T.; Pincus, T.; et al. Serum osteoprotegerin is increased and independently associated with coronary-artery atherosclerosis in patients with rheumatoid arthritis. Atherosclerosis 2007, 195, e135–e141. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Chen, Z.; Jin, Y.; Jiang, B.; Wang, X.; Yu, H.; Yang, X. Incidence and predictors of aorta calcification in patients with systemic lupus erythematosus. Lupus 2019, 28, 275–282. [Google Scholar] [CrossRef]
- Watad, A.; Tiosano, S.; Grysman, N.; Comaneshter, D.; Cohen, A.D.; Shoenfeld, Y.; Amital, H. The association between systemic lupus erythematosus and valvular heart disease: An extensive data analysis. Eur. J. Clin. Investig. 2017, 47, 366–371. [Google Scholar] [CrossRef]
- Plazak, W.; Pasowicz, M.; Kostkiewicz, M.; Podolec, J.; Tomkiewicz-Pajak, L.; Musial, J.; Podolec, P. Influence of chronic inflammation and autoimmunity on coronary calcifications and myocardial perfusion defects in systemic lupus erythematosus patients. Inflamm. Res. 2011, 60, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiani, A.N.; Fishman, E.K.; Petri, M. Aortic valve calcification in systemic lupus erythematosus. Lupus 2006, 15, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Hamano, T.; Obi, Y.; Monden, C.; Oka, T.; Yamaguchi, S.; Matsui, I.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; et al. A Randomized Trial of Magnesium Oxide and Oral Carbon Adsorbent for Coronary Artery Calcification in Predialysis CKD. J. Am. Soc. Nephrol. 2019, 30, 1073–1085. [Google Scholar] [CrossRef]
- Di Iorio, B.; Bellasi, A.; Russo, D.; Investigators, I.S. Mortality in kidney disease patients treated with phosphate binders: A randomized study. Clin. J. Am. Soc. Nephrol. 2012, 7, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakova, T.; Ix, J.H.; Sprague, S.M.; Raphael, K.L.; Fried, L.; Gassman, J.J.; Raj, D.; Cheung, A.K.; Kusek, J.W.; Flessner, M.F.; et al. Rationale and Approaches to Phosphate and Fibroblast Growth Factor 23 Reduction in CKD. J. Am. Soc. Nephrol. 2015, 26, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.D.; Ketteler, M.; Tur, F.; Tur, E.; Isern, B.; Salcedo, C.; Joubert, P.H.; Behets, G.J.; Neven, E.; D’Haese, P.C.; et al. Characterization of SNF472 pharmacokinetics and efficacy in uremic and non-uremic rats models of cardiovascular calcification. PLoS ONE 2018, 13, e0197061. [Google Scholar] [CrossRef] [Green Version]
- Raggi, P.; Bellasi, A.; Bushinsky, D.; Bover, J.; Rodriguez, M.; Ketteler, M.; Sinha, S.; Salcedo, C.; Gillotti, K.; Padgett, C.; et al. Slowing Progression of Cardiovascular Calcification with SNF472 in Patients on Hemodialysis: Results of a Randomized Phase 2b Study. Circulation 2020, 141, 728–739. [Google Scholar] [CrossRef]
- Suzuki, S.; Suzuki, M.; Hanafusa, N.; Tsuchiya, K.; Nitta, K. Denosumab Recovers Aortic Arch Calcification During Long-Term Hemodialysis. Kidney Int. Rep. 2021, 6, 605–612. [Google Scholar] [CrossRef]
- Pawade, T.A.; Doris, M.K.; Bing, R.; White, A.C.; Forsyth, L.; Evans, E.; Graham, C.; Williams, M.C.; van Beek, E.J.R.; Fletcher, A.; et al. Effect of Denosumab or Alendronic Acid on the Progression of Aortic Stenosis: A Double-Blind Randomized Controlled Trial. Circulation 2021, 143, 2418–2427. [Google Scholar] [CrossRef]
- Peng, T.; Zhuo, L.; Wang, Y.; Jun, M.; Li, G.; Wang, L.; Hong, D. Systematic review of sodium thiosulfate in treating calciphylaxis in chronic kidney disease patients. Nephrology 2018, 23, 669–675. [Google Scholar] [CrossRef]
- Djuric, P.; Dimkovic, N.; Schlieper, G.; Djuric, Z.; Pantelic, M.; Mitrovic, M.; Jankovic, A.; Milanov, M.; Kuzmanovic Pficer, J.; Floege, J. Sodium thiosulphate and progression of vascular calcification in end-stage renal disease patients: A double-blind, randomized, placebo-controlled study. Nephrol. Dial. Transpl. 2020, 35, 162–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raggi, P.; Chertow, G.M.; Torres, P.U.; Csiky, B.; Naso, A.; Nossuli, K.; Moustafa, M.; Goodman, W.G.; Lopez, N.; Downey, G.; et al. The ADVANCE study: A randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol. Dial. Transpl. 2011, 26, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giger, E.V.; Castagner, B.; Leroux, J.C. Biomedical applications of bisphosphonates. J. Control. Release 2013, 167, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, S.; Cunningham, J. Is there a role for bisphosphonates in vascular calcification in chronic kidney disease? Bone 2021, 142, 115751. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Neven, E.; Millán, J.L.; Pinkerton, A.B.; D’Haese, P.C.; Verhulst, A. Pharmacological TNAP inhibition efficiently inhibits arterial media calcification in a warfarin rat model but deserves careful consideration of potential physiological bone formation/mineralization impairment. Bone 2020, 137, 115392. [Google Scholar] [CrossRef]
- Tani, T.; Fujiwara, M.; Orimo, H.; Shimizu, A.; Narisawa, S.; Pinkerton, A.B.; Millán, J.L.; Tsuruoka, S. Inhibition of tissue-nonspecific alkaline phosphatase protects against medial arterial calcification and improves survival probability in the CKD-MBD mouse model. J. Pathol. 2020, 250, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millan, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification: A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2022, 118, 84–96. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5, 50. [Google Scholar] [CrossRef] [Green Version]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Fortini, F.; Vieceli Dalla Sega, F.; Marracino, L.; Severi, P.; Rapezzi, C.; Rizzo, P.; Ferrari, R. Well-Known and Novel Players in Endothelial Dysfunction: Updates on a Notch(ed) Landscape. Biomedicines 2021, 9, 997. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Shi, M.; Mosley, J.D.; Weng, C.; Zhang, Y.; Lee, M.T.M.; Jarvik, G.P.; Hakonarson, H.; Namjou-Khales, B.; Sleiman, P.; et al. A Mendelian Randomization Approach Using 3-HMG-Coenzyme-A Reductase Gene Variation to Evaluate the Association of Statin-Induced Low-Density Lipoprotein Cholesterol Lowering with Noncardiovascular Disease Phenotypes. JAMA Netw. Open 2021, 4, e2112820. [Google Scholar] [CrossRef] [PubMed]
- Ference, B.A.; Bhatt, D.L.; Catapano, A.L.; Packard, C.J.; Graham, I.; Kaptoge, S.; Ference, T.B.; Guo, Q.; Laufs, U.; Ruff, C.T.; et al. Association of Genetic Variants Related to Combined Exposure to Lower Low-Density Lipoproteins and Lower Systolic Blood Pressure with Lifetime Risk of Cardiovascular Disease. JAMA 2019, 322, 1381–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Allam, A.H.; Thompson, R.C.; Wann, L.S.; Miyamoto, M.I.; Nur El-Din, A.E.-H.; El-Maksoud, G.A.; Al-Tohamy Soliman, M.; Badr, I.; El-Rahman Amer, H.A.; Sutherland, M.L.; et al. Atherosclerosis in ancient Egyptian mummies: The Horus study. JACC Cardiovasc. Imaging 2011, 4, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.A.; Chen, J.; Nallamshetty, S.; Pham, T.; Goto, S.; Muehlschlegel, J.D.; Libby, P.; Aikawa, M.; Aikawa, E.; Plutzky, J. Retinoids Repress Human Cardiovascular Cell Calcification with Evidence for Distinct Selective Retinoid Modulator Effects. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 656–669. [Google Scholar] [CrossRef]
- Furmanik, M.; van Gorp, R.; Whitehead, M.; Ahmad, S.; Bordoloi, J.; Kapustin, A.; Schurgers, L.J.; Shanahan, C.M. Endoplasmic Reticulum Stress Mediates Vascular Smooth Muscle Cell Calcification via Increased Release of Grp78 (Glucose-Regulated Protein, 78 kDa)-Loaded Extracellular Vesicles. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 898–914. [Google Scholar] [CrossRef]
- Itoh, S.; Mizuno, K.; Aikawa, M.; Aikawa, E. Dimerization of sortilin regulates its trafficking to extracellular vesicles. J. Biol. Chem. 2018, 293, 4532–4544. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.A.; Aikawa, E. Cardiovascular calcification: Artificial intelligence and big data accelerate mechanistic discovery. Nat. Rev. Cardiol. 2019, 16, 261–274. [Google Scholar] [CrossRef] [PubMed]

| Characteristics of |  Valves |  Arteries |
| STRUCTURE | 3 LAYERS: Fibrosa, Spongiosa, Ventricularis | 2 LAYERS: Collagen, Elastin |
| RESIDENT CELLS | FIBROBLAST (VICs) | SMOOTH MUSCLE (VSMCs) |
| PATHOLOGY | DEGENERATIVE | INFLAMMATORY |
| TIME COURSE FOR CALCIFICATION | LONG | SHORT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieceli Dalla Sega, F.; Fortini, F.; Severi, P.; Rizzo, P.; Gardi, I.; Cimaglia, P.; Rapezzi, C.; Tavazzi, L.; Ferrari, R. Cardiac Calcifications: Phenotypes, Mechanisms, Clinical and Prognostic Implications. Biology 2022, 11, 414. https://doi.org/10.3390/biology11030414
Vieceli Dalla Sega F, Fortini F, Severi P, Rizzo P, Gardi I, Cimaglia P, Rapezzi C, Tavazzi L, Ferrari R. Cardiac Calcifications: Phenotypes, Mechanisms, Clinical and Prognostic Implications. Biology. 2022; 11(3):414. https://doi.org/10.3390/biology11030414
Chicago/Turabian StyleVieceli Dalla Sega, Francesco, Francesca Fortini, Paolo Severi, Paola Rizzo, Iija Gardi, Paolo Cimaglia, Claudio Rapezzi, Luigi Tavazzi, and Roberto Ferrari. 2022. "Cardiac Calcifications: Phenotypes, Mechanisms, Clinical and Prognostic Implications" Biology 11, no. 3: 414. https://doi.org/10.3390/biology11030414