1. Introduction
The coronavirus (COVID-19) pandemic is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The pandemic has put millions of people at risk in numerous countries worldwide and caused an unprecedented public health crisis [
1]. Although the origin of COVID-19 (SARS-CoV-2) in humans is still unknown, there are many theories that it could have been transferred to humans from bats [
2]. Likewise, several related coronaviruses (CoVs) have been transmitted from other animals, such as SARS (SARS-CoV) from civets (civets are closely related to cats [
3]), and MERS (MERS-CoV) from dromedary camels [
4]. SARS-CoV-2, and other CoVs, belong to the family coronaviridae (of order nidovirales [
5]), which is a large family of diverse enveloped, positive-sense single-stranded genomic RNA (+ssRNA) viruses that can bring about respiratory diseases in humans and animals [
6]. They are grouped into five genera, namely, alphacoronavirus, betacoronavirus, gammacoronavirus, alphaletovirus, and deltacoronavirus. They infect a range of hosts such as humans, palm civets, bats, dogs, and monkeys, among others [
7]. The alphacoronaviruses and betacoronaviruses mostly infect mammals, and the gammacoronaviruses mostly infect birds. The deltacoronaviruses infect both birds and mammals [
8].
SARS-CoV-2 is the seventh member of the coronavirus family known to affect humans, and the other six are severe acute respiratory syndrome-CoV (SARS-CoV), HCoVs-NL63, HCoVs-OC43, HCoVs-HKU1, HCoVs-229E, and middle east respiratory syndrome-CoV (MERS-CoV) [
9]. SARS-CoV-2 is similar to SARS-CoV, which led to the SARS epidemic in 2003, causing more than 8400 cases and approximately 800 deaths [
10]. Compared to the known SARS-CoV virus, the novel SARS-CoV-2 has a lower mortality rate but a higher human-to-human transmission rate. Similarly, SARS-CoV-2 can have an adverse impact on the human body. It is highly infectious and is a matter of significant concern, since it can not only damage the respiratory system, gastrointestinal system, heart, and central nervous system, but also may lead to multi-organ failure, and eventually, death [
11,
12].
Monitoring zoonotic diseases and host specificity are integral to understanding disease dynamics. Sixty percent of known infectious diseases in humans and 75% of all emerging diseases are zoonotic, as reported by the United Nations Environment Program (UNEP) and the International Livestock Research Institute (ILRI) [
13]. The study of the COVID-19 pandemic is of great significance, not only because it can help healthcare institutions to cope with the ongoing epidemic, but also because it allows researchers to learn more fundamentals about the family coronaviridae, which can provide new knowledge for the prevention of potential pandemics in the future. CoVs are widespread among birds and mammals and can be causes of zoonoses. A zoonosis is defined as any disease or infection that is naturally transmissible from vertebrate animals to humans by the World Health Organization, and COVID-19 has been classified as a zoonotic disease [
14]. One important step to learning about zoonoses and understanding the current pandemic better is finding out how human infections began for SARS-CoV-2. CoVs can lead to various diseases in domestic animals, including dogs, swine, chickens, and cats. Although the origin of COVID-19 in humans is still unknown, genetic analysis results show that it is highly possible that SARS-CoV-2 originates from bats and utilizes the pangolin as an intermediate host [
15,
16,
17].
The CoVs have an envelope membrane that is associated with five structural proteins, namely, the surface (S) protein, or spike protein, haemagglutinin-esterase protein (HE), membrane protein (M), envelope protein (E), and the nucleocapsid protein (N) [
18]; see
Figure 1. The spike protein is responsible for the binding and fusion between the virus and the host cell receptors, and also the infected host cells and adjacent uninfected cells [
19]. The spike protein is further subdivided into two subunits, S1 and S2. The S2 subunit is then again further subdivided into five domains, namely, the fusion peptide (FP), two heptad-repeat regions (HR1 and HR2), the transmembrane domain (TM), and the cytoplasmic tail (CT), all of which play a key role in mediating the viral cell membrane fusion and entry [
20,
21]. Hence, the spike proteins of different CoVs largely determine their ranges of host specificity. Changes in spike protein sequences are reportedly sufficient to change tissue and species tropism and viral virulence [
7,
22,
23]. The S protein is a trimeric transmembrane protein with a protrusion, or spike, on the viral surface, which is the key for binding to and entry into host cells. It is composed of the receptor binding domain or S1 subunit and an S2 subunit that harbor sequences for viral fusion to the cell membrane [
7,
22,
23]. Due to their importance, using the specificity of spike proteins offers an approach to classifying the potential hosts of CoVs.
A common way to classify and understand the dynamics of viruses is to construct a phylogenetic tree of evolution using the sequencing data of the virus [
24,
25]. After the COVID-19 pandemic breakout, databases such as GISAID [
26] collected a large number of sequence data of SARS-CoV-2 from researchers and clinicians worldwide, which can be used for phylogenetic tree inference. Many methods have been developed and applied for constructing phylogenetic trees, including the most similar supertree algorithm (MSSA) method [
27], the MRP method [
28], and the approximate maximum likelihood (ML) supertree method [
29,
30]. Upon similar methods, the state-of-the-art Nextstrain [
24] and IQTREE2 [
25] have been developed. However, these methods of building a phylogenetic tree for CoVs require high computational complexity, and the vast volume of sequence data can cause a scalability issue for phylogeny-based approaches [
31]. For example, Nextstrain [
24] is able to construct trees on thousands of sequences, whereas IQTREE2 [
25] is able to scale to tens of thousands of sequences. There are currently millions of sequences available on GISAID alone—clearly viable alternatives are necessary. Here we study machine learning clustering and classification as an alternative to phylogenetic tree building.
Some efforts have been made to study the coronavirus host data [
32] by using the one-hot embedding (OHE) approach to get fixed length feature embeddings for the spike sequence. OHE provides good predictions, but it has drawbacks, such as the high dimensionality of the feature vectors produced. Furthermore, the columns of the OHE-based vector have a linear relationship, which means that one variable can be easily predicted using the other variables. This behavior can cause parallelism and multicollinearity (when multiple features are correlated with each other) in high dimensions. The authors of [
33,
34] used the coronavirus spike sequences to classify different variants of COVID-19 using
k-mer-based frequency vectors. Researchers have performed clustering on the COVID-19 spike sequences using the same
k-mer-based frequency vector generation approach [
35,
36]. Although their approaches are effective in terms of predictive performance, the dimensionality of the feature vector representation is still high, which can create a very well-known problem in machine learning: the curse of dimensionality. Moreover, for each
k-mer, it is necessary to find the appropriate bin dedicated to a specific
k-mer (“bin matching”) which can be expensive in terms of computational cost.
Another possible solution, which is what we propose here, is the use of the position weight matrix (PWM), sometimes also called a position-specific weight matrix (PSWM) or position-specific scoring matrix (PSSM) [
37]. It is a good representation of motifs in biological sequences. A motif is a nucleotide or amino acid sequence pattern that is widespread and has or is conjectured to have some biological significance. The PWM applies entropy and relative entropy towards identifying transcription factor binding sites (TFBSs), for example. A PWM contains information about the frequencies of nucleotides for each position in the form of weights. These log-odds or log-probability weights are used for computing the binding affinity score.
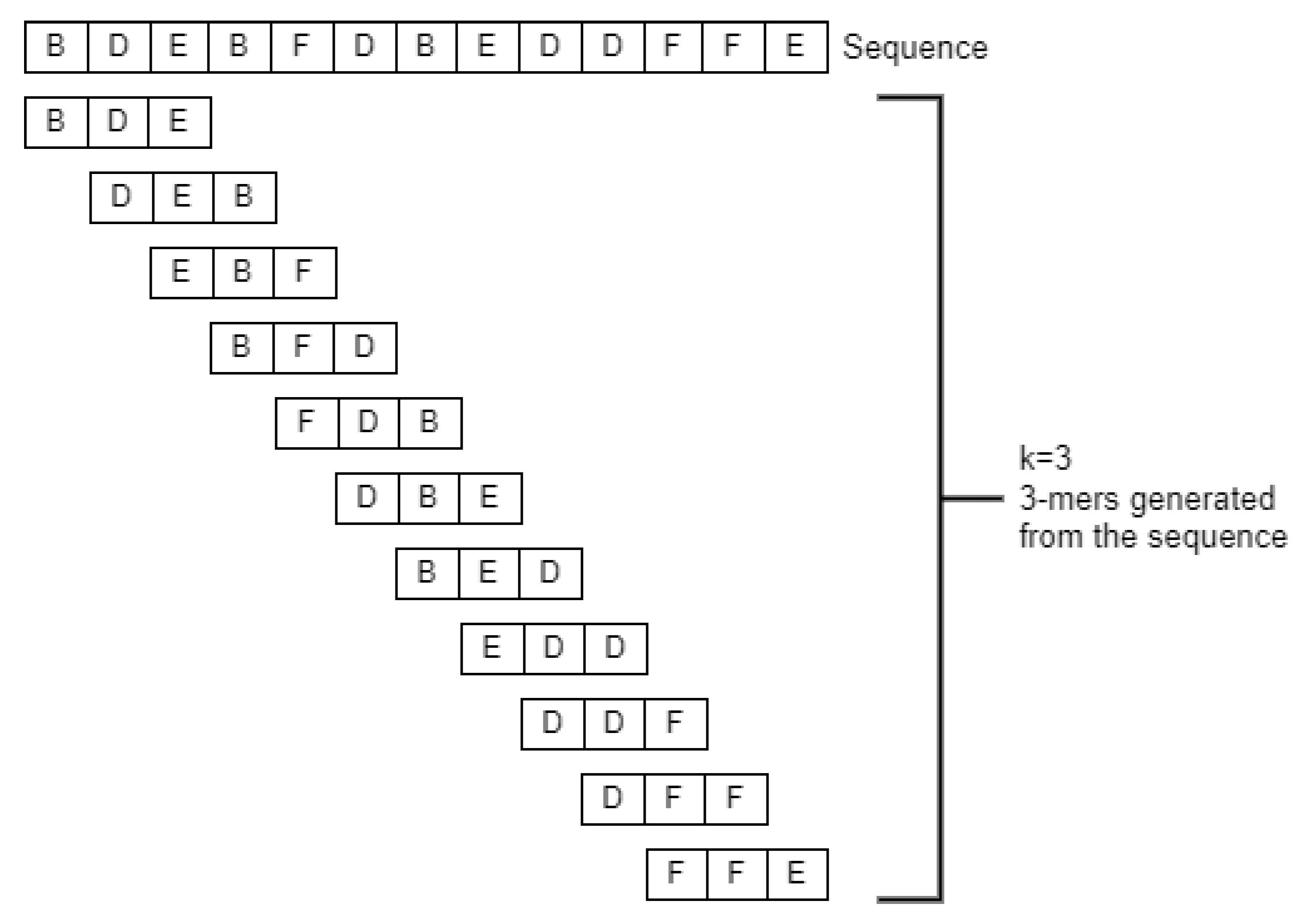
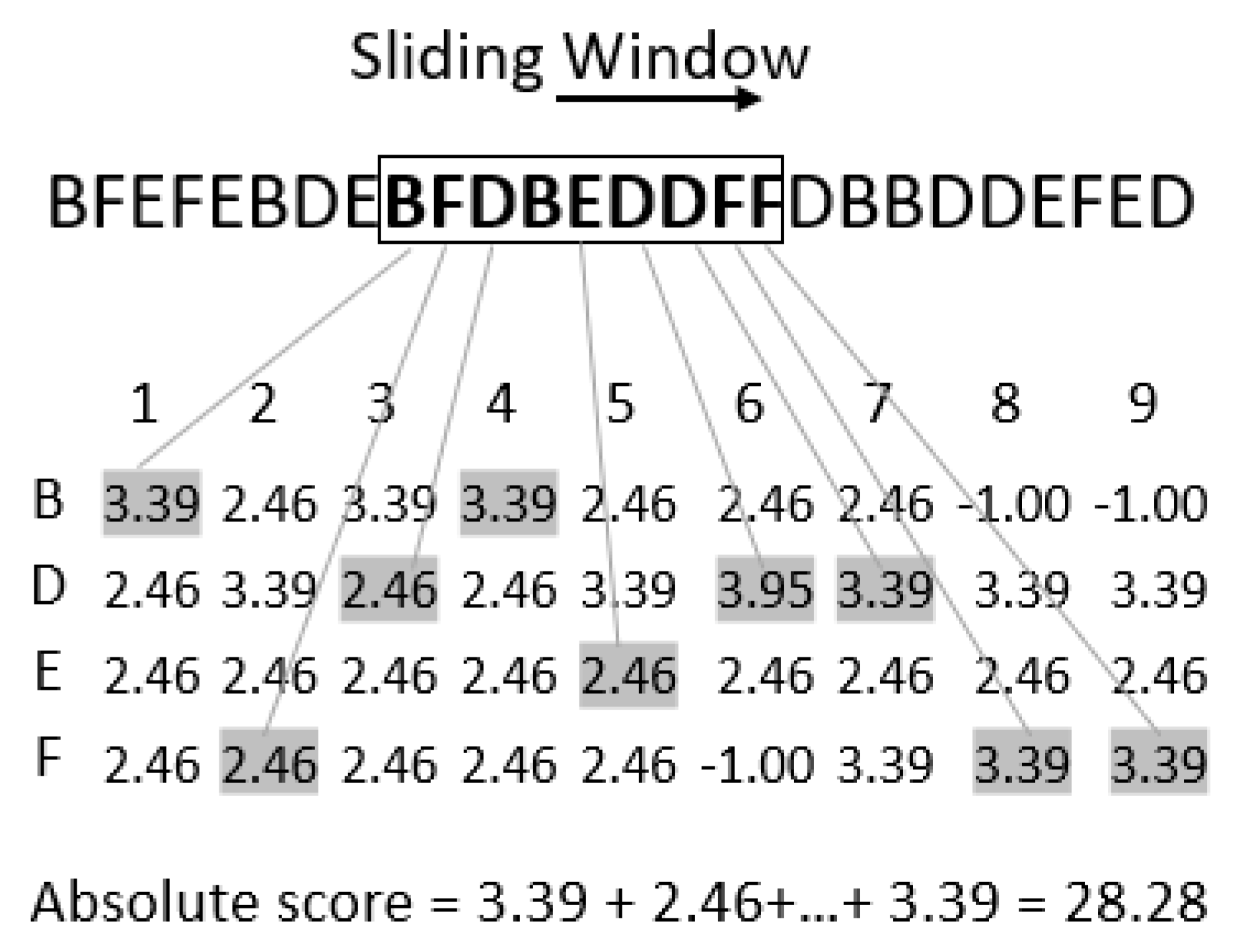
The PWM can be used to distinguish the binding sites from the sequence, a well-known method for de novo motif sequence finding. If we do not know about a possible motif in a given sequence, there are methods such as expectation maximization (EM) and Gibbs sampling, which uses the PWM. Inspired by this application, we computed an absolute score from the PWM while scanning the sequence for “motifs” (here
k-mers) using a sliding window (of size
k, see
Figure 2) and computed the absolute score. We can find relevant information on the motifs based on the score calculated from the PWM. The higher the score, the more relevant the
k-mer is.
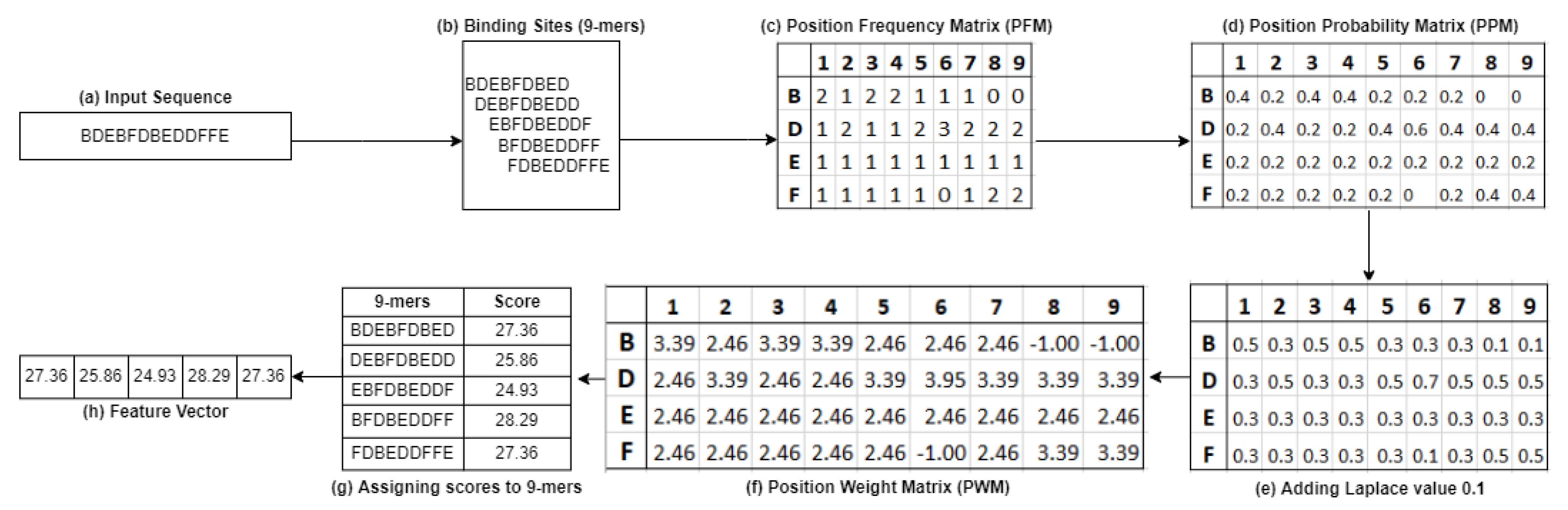
In this paper, we propose an approach called PWM2Vec, a basic implementation of the position weight matrix (PWM) to generate a feature vector representation of a coronavirus spike sequence. Given a spike sequence, we first extract
k-mers. From the
k-mers, we generate the PWM (see
Figure 3). After that, we assign a score to each
k-mer by using the PWM to design a feature embedding and apply machine learning methods such as classification and clustering in this feature embedding. While this is inspired by methods for finding motifs (e.g., TF-promoter binding sites), our goal is to obtain a numerical representation of these
k-mers generated from each sequence.
Our contributions in this paper are as follows:
We propose an approach to generate a fixed-length numerical representation of a spike sequence using a PWM. The generated feature vectors could be used as input to any machine learning algorithm for tasks such as classification and clustering.
Our proposed feature embedding approach contains more compact information and gives better results than the baselines in terms of classification and clustering.
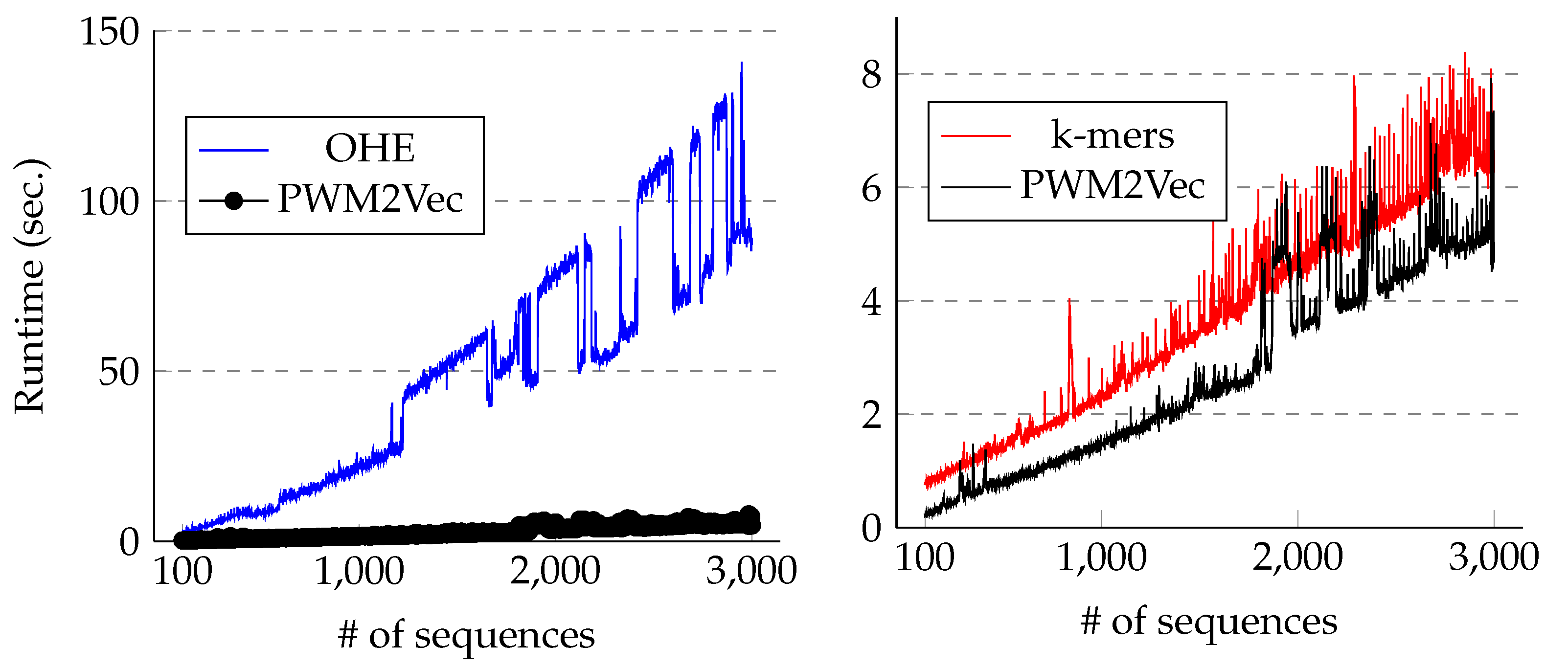
Our feature vector contains fewer dimensions than k-mer and one-hot encoding-based feature vectors (≈20-fold fewer dimensions than one-hot encoding and ≈4-fold fewer dimensions than k-mer-based embedding), which improves the runtimes for classification and clustering algorithms.
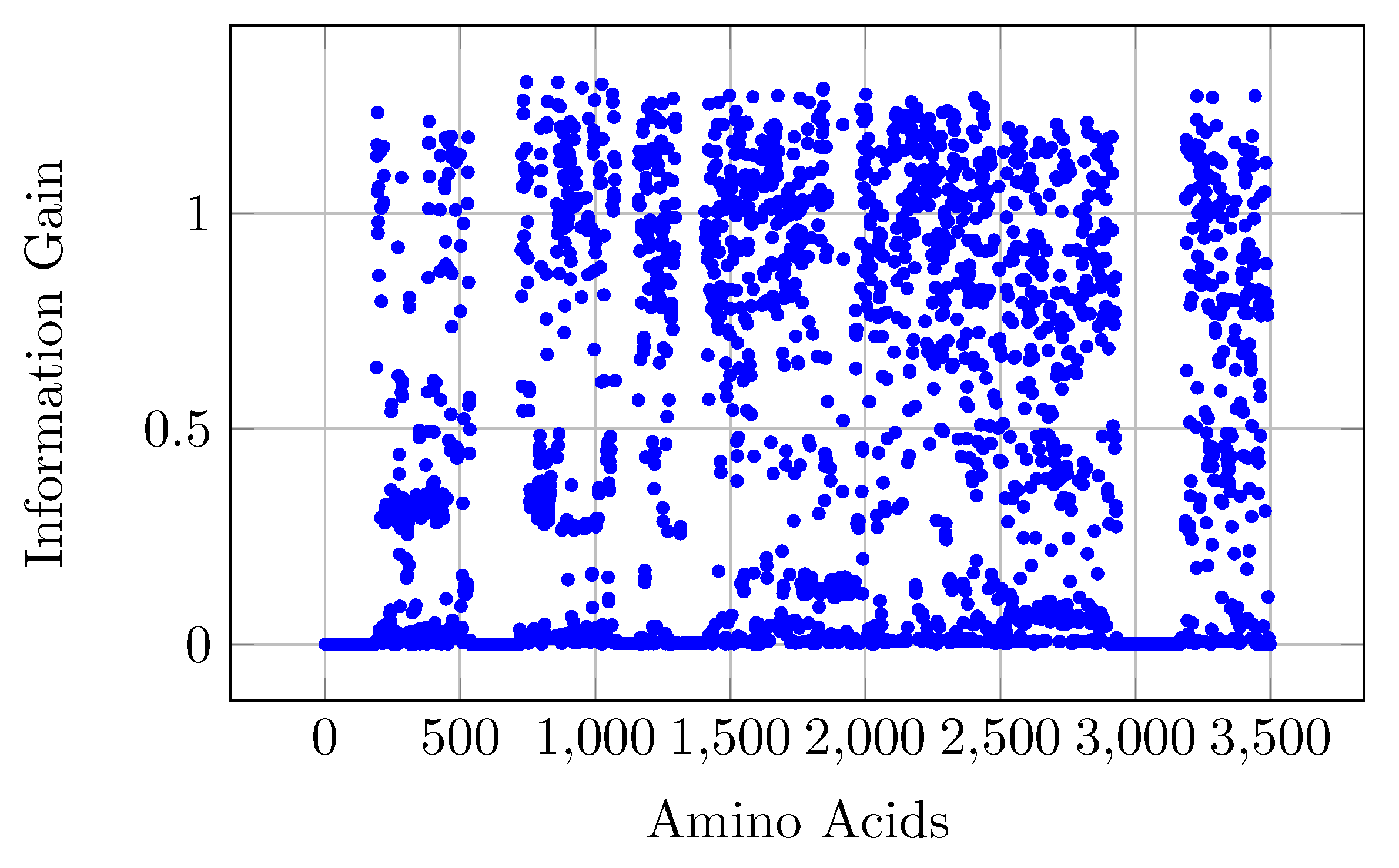
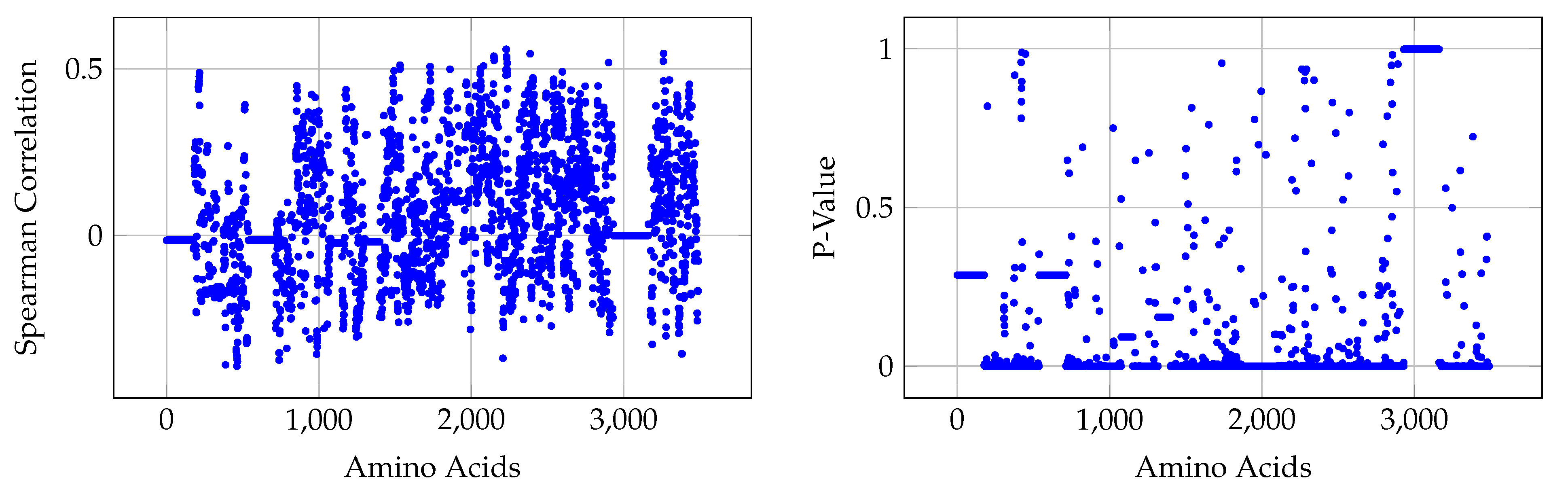
We performed statistical analysis on the data and show the importance of different positions of amino acids that play key roles in the classification of different hosts. We validated the compactness of our proposed embedding from an orthogonal point of view.
The rest of the paper is organized as follows:
Section 2 contains the previous work related to sequence classification, in general, and coronavirus spike sequence classification in particular.
Section 3 contains the details about our proposed alignment-free method for spike sequence classification.
Section 4 contains the experimental setup, dataset collection, and dataset statistics details. The results for our proposed method are given in
Section 5. Finally, we conclude our paper in
Section 6.
2. Related Work
Several machine learning approaches based on
k-mers have been proposed in the literature for classification and clustering tasks [
33,
35,
38,
39]. More specifically, there are many classical algorithms for sequence classification [
40,
41]. Although these methods have been proven to be useful in some studies, it is not clear if they can be used in the context of coronavirus data. Furthermore, another major problem with all those methods is the high computational complexity of the algorithms (because of the high dimensional representation of the data), which can result in higher runtimes for the underlying classification algorithms.
Position weight matrix (PWM)-based approaches have been successfully applied for diverse sequence analysis, motif predictions, and identification studies. Several popular software applications and Web servers have been built based on the implementations of PWMs, e.g., the PWMscan software package [
42] and PSI-BLAST [
43]. Furthermore, many other advanced algorithms have been implemented to optimize PWM-based techniques: examples include MEME (multiple EM for motif elicitation) [
44], based on expectation maximization (EM), and the Gibbs Sampler [
45] for de novo motif discovery, which uses Gibbs sampling algorithms [
46,
47]. The MEME EM algorithm basically finds an initial motif and repeatedly uses EM steps to improve motifs until the PWM values do not improve further [
44,
48]. Furthermore, the BaMM (Bayesian Markov model) algorithm was built based on the Markov algorithm to model correlations among nucleotides at other positions—since the PWM cannot, because the method assumes probabilities at different sites are independent of each other [
47]. The PWM method continues to be applied and extended. Log2PWMs is a simple implementation of PWMs extended to enable conversion or reconstruction of a PWM representation from a sequence logo [
49].
The PWM is also used for the binding specificity of a transcription factor (TF) [
50]. It can be used to scan a sequence for the presence of DNA words, which are comparatively more similar to the PWM than to the background [
51,
52]. Authors in [
53] evaluated the Bayesian network and a support vector machine (SVM) on four different TF binding site-based datasets, and analyzed their performances using PWM. Authors in [
54] developed a tree-based PWM algorithm to simulate the interaction between TF and its binding sites accurately. A new di-nucleotide PWM approach is proposed in [
55] that outperforms the conventional mono-nucleotide PWM method. Moreover, the research done in [
56] proposes an improved position weight matrix (IPWM) method to recognize prokaryotic promoters based on an entropy measurement. Using hepatitis C virus (HCV) nucleotide sequences, the authors of [
57] designed a global PWM for the genotypes of HCV genomes. Then, using the PWM, signatures were selected from the 5’ NCR, CORE, E1, and NS5B regions of the HCV genome. The predictive power of the selected signatures was then evaluated for predicting the most common HCV genotypes and subtypes.
Aside from DNA analysis, the PWM can also be applied to amino acid sequences. Authors in [
58] developed an approach involving the position-specific scoring matrix (PSSM), another name for PWM, to predict protein–protein interactions between protein sequences. First, each protein is transformed into a PSSM, and then the PSSMs are adapted to detect distantly related proteins, the quaternary structural attributes of the proteins, and the proteins’ folding patterns. The research of in [
59] proposes a PWM-based algorithm to predict signal peptide sequences and their cleavage positions in the amino acid sequences of bacteria and eukaryotes. Authors in [
60] developed a PWM-based method for protein function prediction and proposed an argument for why the PWM and associated features have great potential for protein sequence analysis. Although the above methods are successful in their respective domains, they do not provide general means of designing a feature embedding for the underlying sequence, which contains rich information about the sequence and can be used as input to various machine learning algorithms.
The design of efficient feature vector-based representations has been studied in many domains, such as graph analytics [
61,
62], smart grid [
63,
64], electromyography (EMG) [
65], clinical data analysis [
66], network security [
67], and text classification [
68]. After the spread of COVID-19, efforts have been made to study the behavior of the virus using machine learning approaches. Several methods have been proposed recently for the classification of spike sequences. Authors in [
33,
69] used
k-mers along with a kernel-based approach to classify SARS-COV-2 spike sequences. Authors in [
32] proposed the use of one-hot encoding to classify the viral hosts of coronaviridae using spike sequences only. Although they were able to achieve higher predictive performance, some researchers in [
33] showed that the
k-mer-based approach performs better than the one-hot based approach, since it preserves sequence order information more effectively. Efficient clustering of spike sequences was performed in [
35].


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}