
Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies



Abstract
:Simple Summary
Abstract
1. What Are Amyloids?
- Fibrous proteins, such as actin, elastin, and collagen;
- Amyloids, which are associated with many human diseases.
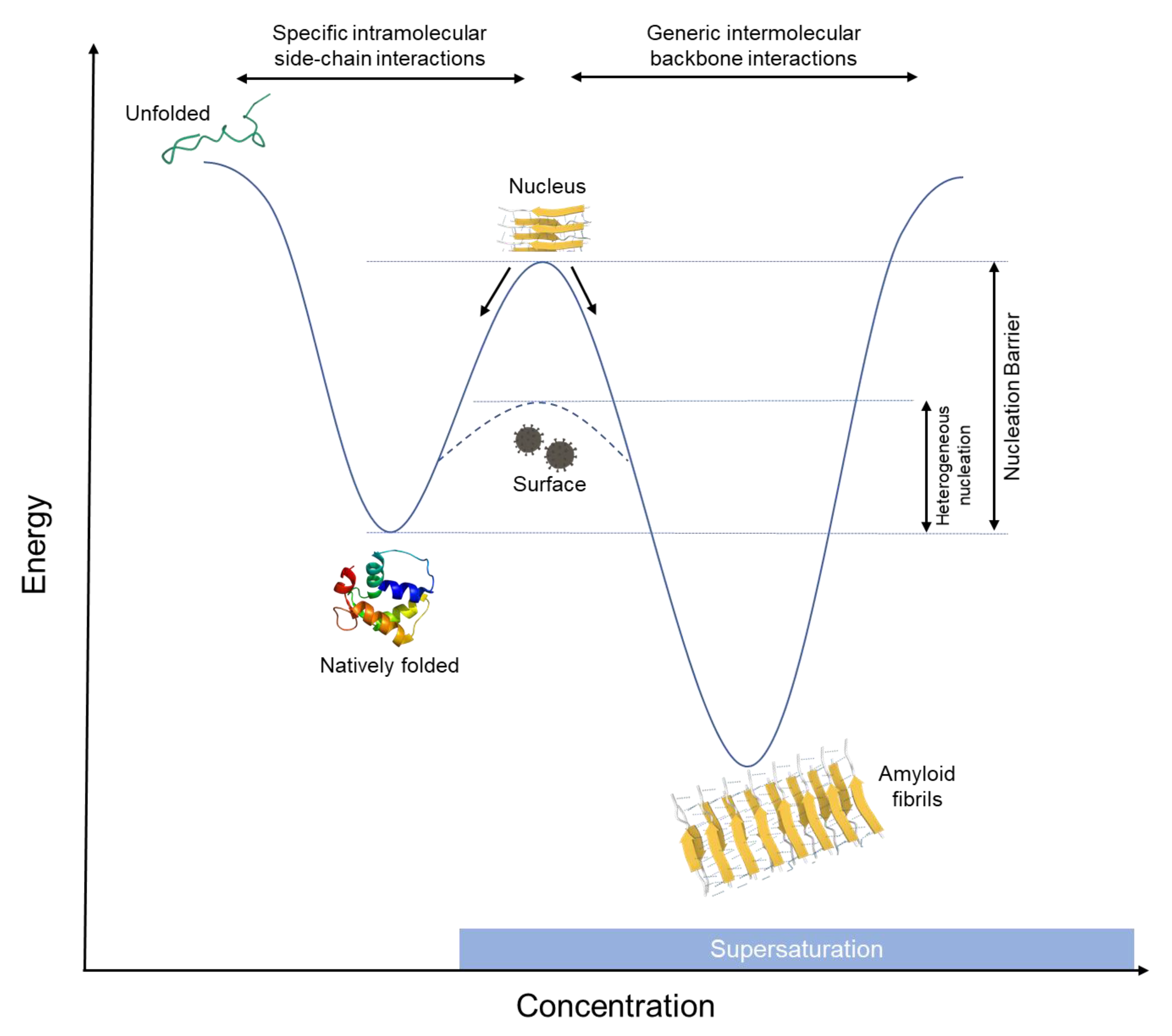
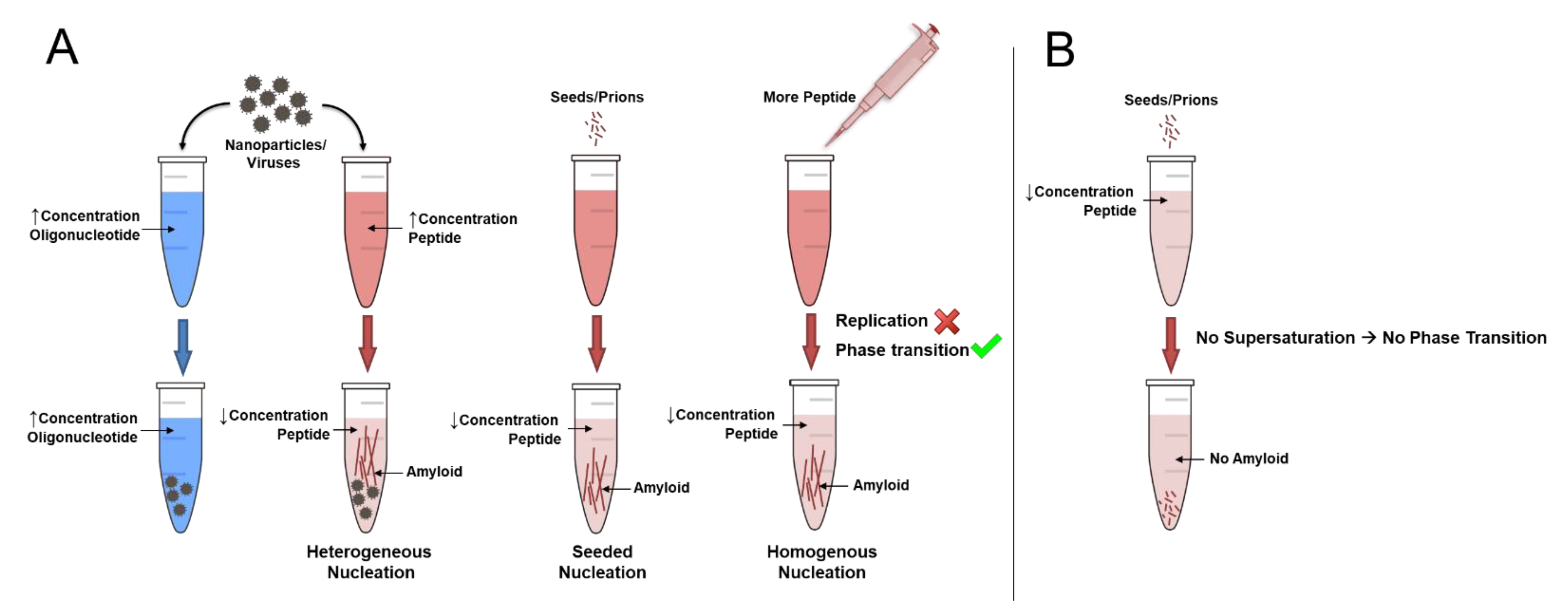
2. How Do Amyloids Form?
3. Can Proteins Replicate?
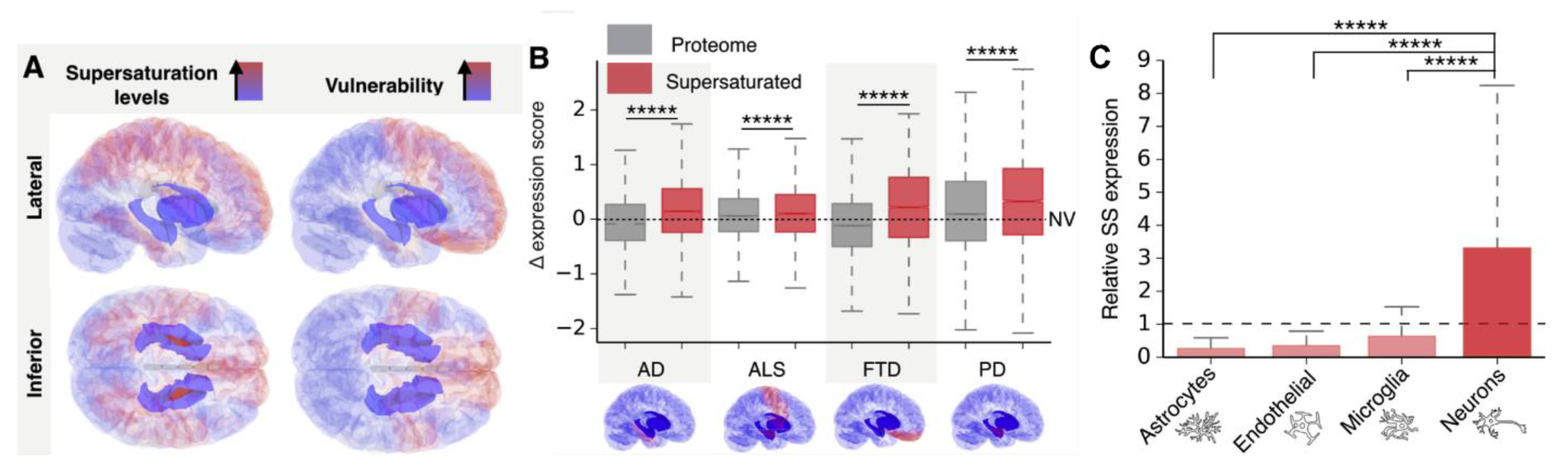
4. How Do Amyloids Cause Toxicity?
5. Conclusions
Funding
Conflicts of Interest
References
- Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozsvar, J.; Yang, C.; Cain, S.A.; Baldock, C.; Tarakanova, A.; Weiss, A.S. Tropoelastin and elastin assembly. Front. Bioeng. Biotechnol. 2021, 9, 138. [Google Scholar] [CrossRef]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [Green Version]
- Fändrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin. Nature 2001, 410, 165–166. [Google Scholar] [CrossRef]
- Jiménez, J.L.; Nettleton, E.J.; Bouchard, M.; Robinson, C.V.; Dobson, C.M.; Saibil, H.R. The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 9196–9201. [Google Scholar] [CrossRef] [Green Version]
- Taboada, P.; Barbosa, S.; Castro, E.; Mosquera, V. Amyloid fibril formation and other aggregate species formed by human serum albumin association. J. Phys. Chem. B 2006, 110, 20733–20736. [Google Scholar] [CrossRef]
- Fändrich, M.; Dobson, C.M. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 2002, 21, 5682–5690. [Google Scholar] [CrossRef] [Green Version]
- Dobson, C.M. The amyloid phenomenon and its links with human disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a023648. [Google Scholar] [CrossRef] [Green Version]
- Ross, E.D.; Edskes, H.K.; Terry, M.J.; Wickner, R.B. Primary sequence independence for prion formation. Proc. Natl. Acad. Sci. USA 2005, 102, 12825–12830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christopher, M. Dobson Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332. [Google Scholar]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.S.; Sawaya, M.R. Structural studies of amyloid proteins at the molecular level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, M.R.; Hughes, M.P.; Rodriguez, J.A.; Riek, R.; Eisenberg, D.S. The expanding amyloid family: Structure, stability, function, and pathogenesis. Cell 2021, 184, 4857–4873. [Google Scholar] [CrossRef]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fa, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef]
- Knowles, T.P.; Fitzpatrick, A.W.; Meehan, S.; Mott, H.R.; Vendruscolo, M.; Dobson, C.M.; Welland, M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science 2007, 318, 1900–1903. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* viewer: Modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Debelouchina, G.T.; Bayro, M.J.; Clare, D.K.; Caporini, M.A.; Bajaj, V.S.; Jaroniec, C.P.; Wang, L.; Ladizhansky, V.; Müller, S.A.; et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. USA 2013, 110, 5468–5473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieleg, O.; Claessens, M.M.A.E.; Bausch, A.R. Structure and dynamics of cross-linked actin networks. Soft Matter 2010, 6, 218–225. [Google Scholar] [CrossRef]
- Fändrich, M.; Schmidt, M.; Grigorieff, N. Recent progress in understanding Alzheimer’s β-amyloid structures. Trends Biochem. Sci. 2011, 36, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petkova, A.T.; Leapman, R.D.; Guo, Z.; Yau, W. Self-propagating, molecular-level polymorphism in Alzheimer’s ß-amyloid fibrils. Science 2005, 307, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Arteni, A. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. eLife 2019, 8, e48907. [Google Scholar] [CrossRef]
- Kresge, N.; Simoni, R.D.; Hill, R.L. The thermodynamic hypothesis of protein folding: The work of Christian Anfinsen. J. Biol. Chem. 2006, 281, e11–e13. [Google Scholar] [CrossRef]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ciryam, P.; Kundra, R.; Morimoto, R.I.; Dobson, C.M.; Vendruscolo, M. Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol. Sci. 2015, 36, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, J.T.; Lansbury, P.T. Seeding “one dimensional cristallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Buell, A.K. The Nucleation of Protein Aggregates—From Crystals to Amyloid Fibrils, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 329, ISBN 9780128122518. [Google Scholar]
- Vekilov, P.G.; Feeling-Taylor, A.R.; Yau, S.T.; Petsev, D. Solvent entropy contribution to the free energy of protein crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 1611–1616. [Google Scholar] [CrossRef] [PubMed]
- Vorontsova, M.A.; Vekilov, P.G. Recent advances in the understanding of two-step nucleation of protein crystals. Faraday Discuss. 2015, 179, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, P.G. Kinetics and mechanisms of protein crystallization at the molecular level. Methods Mol. Biol. 2005, 300, 15–52. [Google Scholar] [CrossRef] [PubMed]
- Eaton, W.A.; Muñoz, V.; Thompson, P.A.; Chan, C.K.; Hofrichter, J. Submillisecond kinetics of protein folding. Curr. Opin. Struct. Biol. 1997, 7, 10–14. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. The amyloid as a ribbon-like micelle in contrast to spherical micelles represented by globular proteins. Molecules 2019, 24, 4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikenoue, T.; Lee, Y.H.; Kardos, J.; Yagi, H.; Ikegami, T.; Naiki, H.; Goto, Y. Heat of supersaturation-limited amyloid burst directly monitored by isothermal titration calorimetry. Proc. Natl. Acad. Sci. USA 2014, 111, 6654–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, M.; Lin, Y.; Dai, I.; Okumura, M.; Markova, N.; Ladbury, J.E.; Sterpone, F.; Lee, Y.H. Energy landscape of polymorphic amyloid generation of β2-microglobulin revealed by calorimetry. Chem. Commun. 2018, 54, 7995–7998. [Google Scholar] [CrossRef]
- Karthika, S.; Radhakrishnan, T.K.; Kalaichelvi, P. A review of classical and nonclassical nucleation theories. Cryst. Growth Des. 2016, 16, 6663–6681. [Google Scholar] [CrossRef]
- Vekilov, P.G. Nucleation. Cryst. Growth Des. 2010, 10, 5007–5019. [Google Scholar] [CrossRef]
- De Yoreo, J.J. Principles of Crystal Nucleation and Growth. Rev. Mineral. Geochem. 2003, 54, 57–93. [Google Scholar] [CrossRef] [Green Version]
- Kamano, Y.; Kadota, K.; Shimosaka, A.; Shirakawa, Y.; Hidaka, J. Quantitative evaluation on the heterogeneous nucleation of amino acid by a thermodynamic analysis. J. Mol. Liq. 2014, 200, 474–479. [Google Scholar] [CrossRef]
- Hu, G.; Cai, X.; Rong, Y. Phase Transformation and Properties; De Gruyter: Berlin, Germany, 2021. [Google Scholar]
- Habchi, J.; Chia, S.; Galvagnion, C.; Michaels, T.C.T.; Bellaiche, M.M.J.; Ruggeri, F.S.; Sanguanini, M.; Idini, I.; Kumita, J.R.; Sparr, E.; et al. Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat. Chem. 2018, 10, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, M.S.; Yagi, H.; Adachi, M.; Lee, Y.H.; Goto, Y. Small liposomes accelerate the fibrillation of amyloid beta (1–40). J. Biol. Chem. 2015, 290, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terakawa, M.S.; Lin, Y.; Kinoshita, M.; Kanemura, S.; Itoh, D.; Sugiki, T.; Okumura, M.; Ramamoorthy, A.; Lee, Y.-H. Impact of membrane curvature on amyloid aggregation. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 1741–1764. [Google Scholar] [CrossRef]
- Knight, J.D.; Miranker, A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004, 341, 1175–1187. [Google Scholar] [CrossRef]
- Linse, S.; Cabaleiro-Lago, C.; Xue, W.-F.; Lynch, I.; Lindman, S.; Thulin, E.; Radford, S.E.; Dawson, K. A Nucleation of protein fibrillation by nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 8691–8696. [Google Scholar] [CrossRef] [Green Version]
- Gladytz, A.; Abel, B.; Risselada, H.J. Gold-induced fibril growth: The mechanism of surface-facilitated amyloid aggregation. Angew. Chem. Int. Ed. 2016, 55, 11242–11246. [Google Scholar] [CrossRef]
- Gladytz, A.; Wagner, M.; Häupl, T.; Elsner, C.; Abel, B. Structure-making effects of metal nanoparticles in amyloid peptide fibrillation. Part. Part. Syst. Charact. 2015, 32, 573–582. [Google Scholar] [CrossRef]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; Thompson, E.A.; et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef] [Green Version]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. The effect of glycosaminoglycans (GAGs) on amyloid aggregation and toxicity. Molecules 2015, 20, 2510–2528. [Google Scholar] [CrossRef] [Green Version]
- Cohlberg, J.A.; Li, J.; Uversky, V.N.; Fink, A.L. Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from α-synuclein in vitro. Biochemistry 2002, 41, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y. Nucleic Acid-Mediated Protein Aggregation and Assembly, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 84, ISBN 9780123864833. [Google Scholar]
- Tö, M.; Michaels, T.C.T.; Sanagavarapu, K.; Yang, X.; Meisl, G.; Cohen, S.I.A.; Knowles Bd, T.P.J.; Linse, S. Secondary nucleation in amyloid formation. Chem. Commun. 2018, 54, 8667–8684. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Movafaghi, S.; Francino Urdániz, I.M.; Rowe, T.M.; Goodwin, A.; Randolph, T.W. Insulin fibril formation caused by mechanical shock and cavitation. J. Phys. Chem. B 2021, 125, 8021–8027. [Google Scholar] [CrossRef]
- Malik, R.; Roy, I. Probing the mechanism of insulin aggregation during agitation. Int. J. Pharm. 2011, 413, 73–80. [Google Scholar] [CrossRef]
- Koloteva-Levine, N.; Aubrey, L.D.; Marchante, R.; Purton, T.J.; Hiscock, J.R.; Tuite, M.F.; Xue, W.F.; Jennifer, R. Amyloid particles facilitate surface-catalyzed cross-seeding by acting as promiscuous nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2104148118. [Google Scholar] [CrossRef]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Firat-Karalar, E.N.; Welch, M.D. New mechanisms and functions of actin nucleation. Curr. Opin. Cell Biol. 2011, 23, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Salsas-Escat, R.; Stultz, C.M. The molecular mechanics of collagen degradation: Implications for human disease. Exp. Mech. 2009, 49, 65–77. [Google Scholar] [CrossRef]
- Plastino, J.; Blanchoin, L. Dynamic stability of the actin ecosystem. J. Cell Sci. 2019, 132, jcs219832. [Google Scholar] [CrossRef] [Green Version]
- Rauscher, S.; Baud, S.; Miao, M.; Keeley, F.W.W.; Pomès, R. Proline and glycine control protein self-organization into elastomeric or amyloid fibrils. Structure 2006, 14, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.A.; Rubenstein, E. Proline: The Distribution, Frequency, Positioning, and Common Functional Roles of Proline and Polyproline Sequences in the Human Proteome. PLoS ONE 2013, 8, 53785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguzzi, A.; De Cecco, E. Shifts and drifts in prion science. Science 2020, 370, 32–34. [Google Scholar] [CrossRef]
- Malmberg, M.; Malm, T.; Gustafsson, O.; Wright, A.; El Andaloussi, S.; Ezzat, K. Disentangling the amyloid pathways: A mechanistic approach to etiology. Front. Neurosci. 2020, 14, 256. [Google Scholar] [CrossRef] [Green Version]
- Freer, R.; Sormanni, P.; Ciryam, P.; Rammner, B.; Rizzoli, S.O.; Dobson, C.M.; Vendruscolo, M. Supersaturated proteins are enriched at synapses and underlie cell and tissue vulnerability in Alzheimer’s disease. Heliyon 2019, 5, e02589. [Google Scholar] [CrossRef]
- Freer, R.; Sormanni, P.; Vecchi, G.; Ciryam, P.; Dobson, C.M.; Vendruscolo, M. A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer’s disease. Sci. Adv. 2016, 2, e1600947. [Google Scholar] [CrossRef] [Green Version]
- Porcari, R.; Proukakis, C.; Waudby, C.A.; Bolognesi, B.; Mangione, P.P.; Paton, J.F.S.; Mullin, S.; Cabrita, L.D.; Penco, A.; Relini, A.; et al. The H50Q mutation induces a 10-fold decrease in the solubility of α-synuclein. J. Biol. Chem. 2015, 290, 2395–2404. [Google Scholar] [CrossRef] [Green Version]
- Fagan, A.M.; Henson, R.L.; Li, Y.; Boerwinkle, A.H.; Xiong, C.; Bateman, R.J.; Goate, A.; Ances, B.M.; Doran, E.; Christian, B.T.; et al. Comparison of CSF biomarkers in down syndrome and autosomal dominant Alzheimer’s disease: A cross-sectional study. Lancet Neurol. 2021, 20, 615–626. [Google Scholar] [CrossRef]
- Kasuga, K.; Tokutake, T.; Ishikawa, A.; Uchiyama, T.; Tokuda, T.; Onodera, O.; Nishizawa, M.; Ikeuchi, T. Differential levels of α-synuclein, β-amyloid42 and tau in CSF between patients with dementia with Lewy bodies and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2010, 81, 608–610. [Google Scholar] [CrossRef]
- D’Aguanno, V.; Ralli, M.; Artico, M.; Russo, F.Y.; Scarpa, A.; Fiore, M.; Tirassa, P.; Severini, C.; de Vincentiis, M.; Greco, A. Systemic amyloidosis: A contemporary overview. Clin. Rev. Allergy Immunol. 2019, 59, 304–322. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Dispenzieri, A. Systemic amyloidosis recognition, prognosis, and therapy: A systematic review. JAMA 2020, 324, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.B.; Inoue, Y.; Mehra, M.R. Amyloidosis and the heart: A comprehensive review. Arch. Intern. Med. 2006, 166, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Surin, A.K.; Grishin, S.Y.; Galzitskaya, O.V. Determination of amyloid core regions of insulin analogues fibrils. Prion 2020, 14, 149–162. [Google Scholar] [CrossRef]
- Nagase, T.; Iwaya, K.; Iwaki, Y.; Kotake, F.; Uchida, R.; Oh-I, T.; Sekine, H.; Miwa, K.; Murakami, S.; Odaka, T.; et al. Insulin-derived amyloidosis and poor glycemic control: A case series. Am. J. Med. 2014, 127, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.L.; Gallo, C.V.D.M.; Costa, D.C.F.; Rangel, L.P.; De Moura Gallo, C.V.; Costa, D.C.F.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like p53 amyloids in cancer. Biochemistry 2019, 59, 146–155. [Google Scholar] [CrossRef]
- Liebman, S.W.; Chernoff, Y.O. Prions in yeast. Genetics 2012, 191, 1041–1072. [Google Scholar] [CrossRef] [Green Version]
- Wickner, R.B.; Edskes, H.K.; Son, M.; Bezsonov, E.E.; DeWilde, M.; Ducatez, M. Yeast prions compared to functional prions and amyloids. J. Mol. Biol. 2018, 430, 3707–3719. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Kryndushkin, D.; Wickner, R.B. Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. USA 2011, 108, 5337–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denroche, H.C.; Verchere, C.B. IAPP and type 1 diabetes: Implications for immunity, metabolism and islet transplants. J. Mol. Endocrinol. 2018, 60, R57–R75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chételat, G.; La Joie, R.; Villain, N.; Perrotin, A.; De La Sayette, V.; Eustache, F.; Vandenberghe, R. Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. NeuroImage Clin. 2013, 2, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Sturchio, A.; Dwivedi, A.K.; Young, C.B.; Malm, T.; Marsili, L.; Sharma, J.S.; Mahajan, A.; Hill, E.J.; El Andaloussi, S.; Poston, K.L.; et al. High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis. EClinicalMedicine 2021, 38, 100988. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Amyloid b-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef]
- Dear, A.J.; Michaels, T.C.T.; Meisl, G.; Klenerman, D.; Wu, S.; Perrett, S.; Linse, S.; Dobson, C.M.; Knowles, T.P.J. Kinetic diversity of amyloid oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 201922267. [Google Scholar] [CrossRef]
- Sancesario, G.M.; Cencioni, M.T.; Esposito, Z.; Borsellino, G.; Nuccetelli, M.; Martorana, A.; Battistini, L.; Sorge, R.; Spalletta, G.; Ferrazzoli, D.; et al. The load of amyloid-β oligomers is decreased in the cerebrospinal fluid of Alzheimer’s disease patients. J. Alzheimers Dis. 2012, 31, 865–878. [Google Scholar] [CrossRef]
- Jongbloed, W.; Bruggink, K.A.; Kester, M.I.; Visser, P.J.; Scheltens, P.; Blankenstein, M.A.; Verbeek, M.M.; Teunissen, C.E.; Veerhuis, R. Amyloid-β oligomers relate to cognitive decline in alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 35–43. [Google Scholar] [CrossRef]
- Van Helmond, Z.; Miners, J.S.; Kehoe, P.G.; Love, S. Higher soluble amyloid β concentration in frontal cortex of young adults than in normal elderly or Alzheimer’s disease. Brain Pathol. 2010, 20, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. When loss is gain: Reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007, 8, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Southam, K.A.; Stennard, F.; Pavez, C.; Small, D.H. Knockout of amyloid β protein precursor (APP) expression alters synaptogenesis, neurite branching and axonal morphology of hippocampal neurons. Neurochem. Res. 2019, 44, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Fa, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M.; et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 2011, 69, 819–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benskey, M.J.; Sellnow, R.C.; Sandoval, I.M.; Sortwell, C.E.; Lipton, J.W.; Manfredsson, F.P. Silencing alpha synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front. Mol. Neurosci. 2018, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In vivo RNAi-mediated α-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Collier, T.J.; Eugene Redmond, D.; Steece-Collier, K.; Lipton, J.W.; Manfredsson, F.P. Is alpha-synuclein loss-of-function a contributor to parkinsonian pathology? Evidence from non-human primates. Front. Neurosci. 2016, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, R.; Ferreira, E.; Tran, A.; Turner, E.C.; Belfiore, R.; Branca, C.; Oddo, S. Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell 2018, 17, e12775. [Google Scholar] [CrossRef] [Green Version]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.A.; Weis, J.; et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, 310–318. [Google Scholar] [CrossRef]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.A.; Fisher, E.M.C.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Velde, C.V.; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2009, 19, 671–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espay, A.J.; Sturchio, A.; Schneider, L.S.; Ezzat, K. Soluble Amyloid-β Consumption in Alzheimer’s Disease. J. Alzheimers Dis. 2021, 82, 1403–1415. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fibrous Proteins | Amyloid Fibrils |
|---|---|
| Specific proteins | Any protein sequence |
| Monomers assemble in their native conformation via specific intramolecular sidechain-based interactions | Proteins assemble into in cross-β conformation via generic intermolecular backbone interactions |
| Functional domains remain accessible | Majority of functional domains are buried in steric zippers |
| Form well-defined networks | Precipitate into plaques |
| Controlled nucleation and growth via structural elements (proline and glycine rich), capping proteins, specific nucleators, enzymes and ATP | Uncontrolled |
| Reversible | Irreversible |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ezzat, K.; Sturchio, A.; Espay, A.J. Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies. Biology 2022, 11, 535. https://doi.org/10.3390/biology11040535
Ezzat K, Sturchio A, Espay AJ. Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies. Biology. 2022; 11(4):535. https://doi.org/10.3390/biology11040535
Chicago/Turabian StyleEzzat, Kariem, Andrea Sturchio, and Alberto J. Espay. 2022. "Proteins Do Not Replicate, They Precipitate: Phase Transition and Loss of Function Toxicity in Amyloid Pathologies" Biology 11, no. 4: 535. https://doi.org/10.3390/biology11040535