
Distance-Based Phylogenetic Placement with Statistical Support

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Support Estimation Methods
2.1.1. Nonparametric Bootstrapping
Branch Length Re-Estimation
Linear Algebraic Formulation and Implementation
2.1.2. Parametric Sampling (Binomial and Poisson)
2.1.3. Nonparametric Subsampling
2.2. Experimental Setup
2.2.1. Methods
Distance Calculations
2.2.2. Datasets
Simulated Single-Gene RNASim
SEPP Simulated Fragmentary Dataset
Web of Life (WOL)
2.2.3. Evaluation Criteria
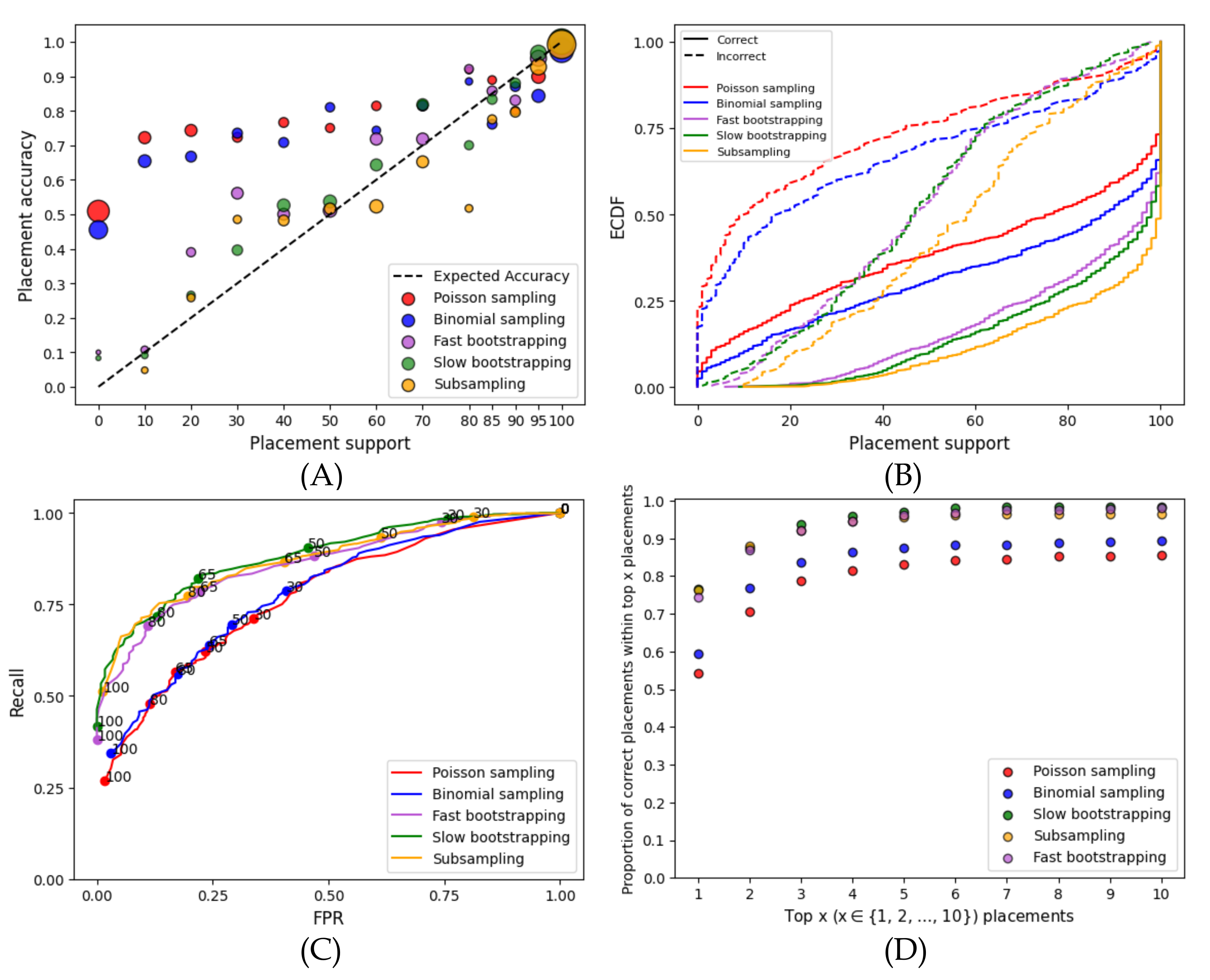
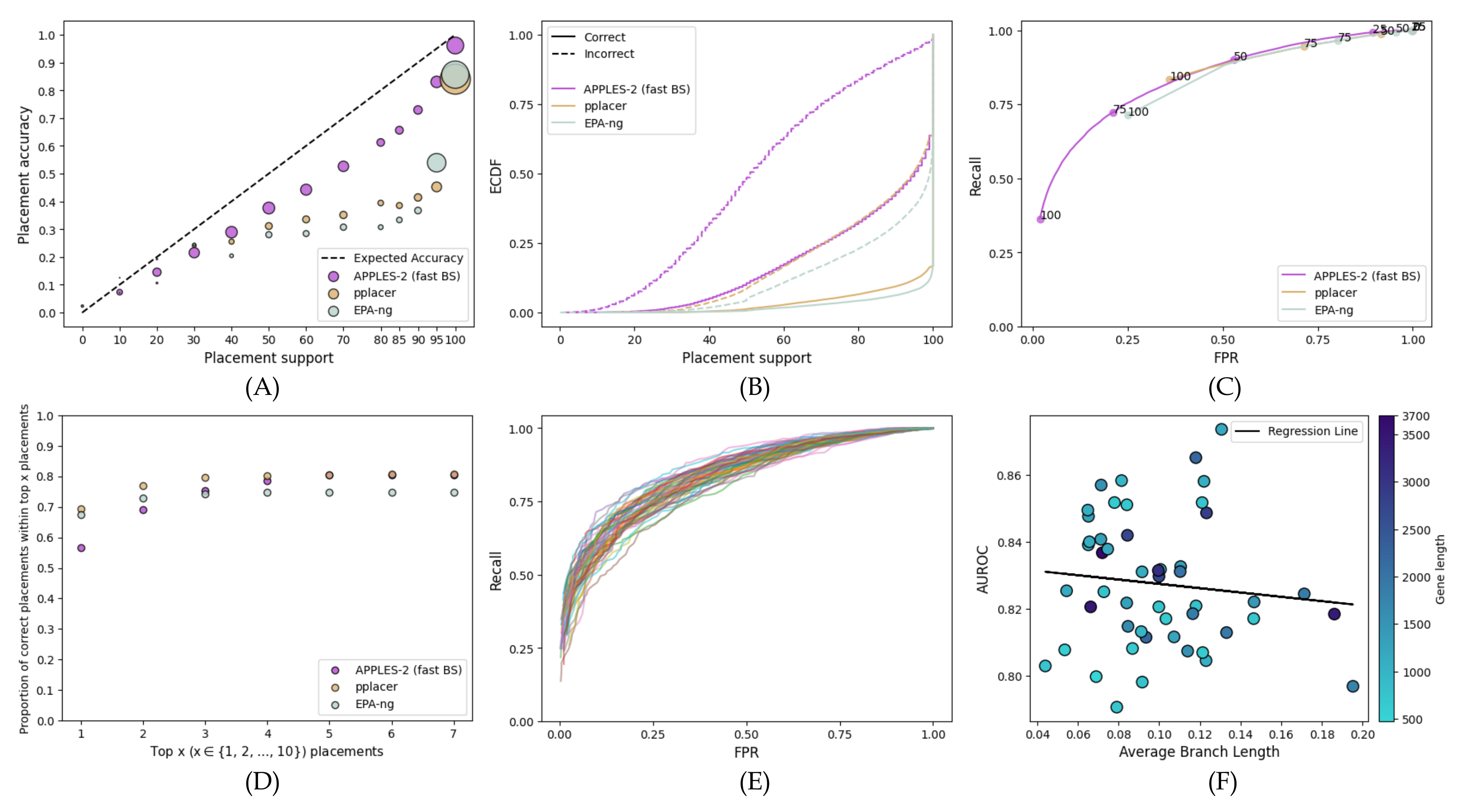
- Calibration: We first bin branches by their support into several groups and quantify the relationship between bins of branch support and the percentage of correctly placed queries in each bin. For example, for branches in the 40–50% support bin, we say the results are calibrated if roughly 45% of these branches are correct. When discussing calibration, we also report the mean squared error (MSE) between the observed and expected accuracies; Lower MSE values indicate that the method is more calibrated.
- Predictive power (ROC): We ask if support values can effectively distinguish correct from incorrect branches using receiver operating characteristic (ROC) curves, which depict the relationship between the percentage of all true branches with support that lie above some threshold T (recall), and the percentage of all false branches with support that lie below T (false-positive rate; FPR). For , we label each correct branch with support s as if and as if , and we label each incorrect branch as if , and as if . We then plot versus .
- Empirical Cumulative Distribution Function (ECDF): Another way to examine support values is to study their ECDF, separating the correct and incorrect branches. Ideally, incorrect branches have low support (uniformly distributed), and correct branches have high support (depending on the signal, and hence, the power). Generally, a wider difference between the distribution of correct and incorrect branches is desired.
3. Results
3.1. Alternative Support Estimation Methods for APPLES-2
3.2. Comparison with Existing ML Methods
3.2.1. Full-Length Single-Gene Simulated Data
3.2.2. Fragmentary Single-Gene Simulated Data
Fragmentary RNASim Dataset
Fragmentary SEPP Dataset
3.2.3. Multi-Gene Web of Life (WOL) Real Dataset
Single-Gene Placement on the Gene Tree
Discordant Placement
Multi-Gene Concatenation
3.3. Running Time
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ML | Maximum Likelihood |
| JC | Jukes–Cantor |
| WOL | Web Of Life (dataset) |
| ROC | Receiver Operating Characteristic |
| FPR | False Positive Rate |
| AUROC | Area Under Receiver Operating Characteristic |
| MSE | Mean Squared Error |
| LSE | Least Squared Error |
| ECDF | Empirical Commulative Distribution Function |
Appendix A. Additional Data
Additional Figures




References
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 2018, 3, 00021-18. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A. Phylogenetics and the Human Microbiome. Syst. Biol. 2015, 64, e26–e41. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.; Evans, S.N. Edge Principal Components and Squash Clustering: Using the Special Structure of Phylogenetic Placement Data for Sample Comparison. PLoS ONE 2013, 8, e56859. [Google Scholar] [CrossRef]
- Nguyen, N.P.; Mirarab, S.; Liu, B.; Pop, M.; Warnow, T. TIPP: Taxonomic identification and phylogenetic profiling. Bioinformatics 2014, 30, 3548–3555. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Asnicar, F.; Thomas, A.M.; Beghini, F.; Mengoni, C.; Manara, S.; Manghi, P.; Zhu, Q.; Bolzan, M.; Cumbo, F.; May, U.; et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 2020, 11, 2500. [Google Scholar] [CrossRef]
- Darling, A.E.; Jospin, G.; Lowe, E.; Matsen, F.A.; Bik, H.M.; Eisen, J.A. PhyloSift: Phylogenetic analysis of genomes and metagenomes. PeerJ 2014, 2, e243. [Google Scholar] [CrossRef]
- Bohmann, K.; Mirarab, S.; Bafna, V.; Gilbert, M.T.P. Beyond DNA barcoding: The unrealized potential of genome skim data in sample identification. Mol. Ecol. 2020, 29, 2521–2534. [Google Scholar] [CrossRef]
- Balaban, M.; Sarmashghi, S.; Mirarab, S. APPLES: Scalable Distance-Based Phylogenetic Placement with or without Alignments. Syst. Biol. 2020, 69, 566–578. [Google Scholar] [CrossRef]
- Libin, P.; Eynden, E.V.; Incardona, F.; Nowé, A.; Bezenchek, A.; Sönnerborg, A.; Vandamme, A.M.; Theys, K.; Baele, G. PhyloGeoTool: Interactively exploring large phylogenies in an epidemiological context. Bioinformatics 2017, 33, 3993–3995. [Google Scholar] [CrossRef]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2019, 68, 365–369. [Google Scholar] [CrossRef]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010, 11, 538. [Google Scholar] [CrossRef]
- Stark, M.; Berger, S.A.; Stamatakis, A.; von Mering, C. MLTreeMap—Accurate Maximum Likelihood placement of environmental DNA sequences into taxonomic and functional reference phylogenies. BMC Genom. 2010, 11, 461. [Google Scholar] [CrossRef]
- Wedell, E.; Cai, Y.; Warnow, T. Scalable and Accurate Phylogenetic Placement Using pplacer-XR. In Proceedings of the Algorithms for Computational Biology, Missoula, MT, USA, 7–11 June 2021; AlCoB 2021, Lecture Notes in Computer Science. Springer: Cham, Switzerland, 2021; pp. 94–105. [Google Scholar] [CrossRef]
- Balaban, M.; Jiang, Y.; Roush, D.; Zhu, Q.; Mirarab, S. Fast and accurate distance-based phylogenetic placement using divide and conquer. Mol. Ecol. Resour. 2022, 22, 1213–1227. [Google Scholar] [CrossRef]
- Balaban, M.; Mirarab, S. Phylogenetic double placement of mixed samples. Bioinformatics 2020, 36, i335–i343. [Google Scholar] [CrossRef]
- Linard, B.; Swenson, K.M.; Pardi, F. Rapid alignment-free phylogenetic identification of metagenomic sequences. Bioinformatics 2019, 35, 3303–3312. [Google Scholar] [CrossRef]
- Brown, D.; Truszkowski, J. LSHPlace: Fast phylogenetic placement using locality-sensitive hashing. In Proceedings of the Pacific Symposium On Biocomputing, Kohala Coast, HI, USA, 3–7 January 2013; pp. 310–319. [Google Scholar] [CrossRef]
- Jiang, Y.; Balaban, M.; Zhu, Q.; Mirarab, S. DEPP: Deep Learning Enables Extending Species Trees using Single Genes. Syst. Biol. 2022. [Google Scholar] [CrossRef]
- Jiang, Y.; Tabaghi, P.; Mirarab, S. Phylogenetic Placement Problem: A Hyperbolic Embedding Approach. In Proceedings of the Comparative Genomics, La Jolla, CA, USA, 20–21 May 2022; Jin, L., Durand, D., Eds.; Springer International Publishing: Cham, Switzerland, 2022; pp. 68–85. [Google Scholar] [CrossRef]
- Zheng, Q.; Bartow-McKenney, C.; Meisel, J.S.; Grice, E.A. HmmUFOtu: An HMM and phylogenetic placement based ultra-fast taxonomic assignment and OTU picking tool for microbiome amplicon sequencing studies. Genome Biol. 2018, 19, 82. [Google Scholar] [CrossRef]
- Rabiee, M.; Mirarab, S. INSTRAL: Discordance-Aware Phylogenetic Placement Using Quartet Scores. Syst. Biol. 2020, 69, 384–391. [Google Scholar] [CrossRef]
- Mai, U.; Mirarab, S. Completing gene trees without species trees in sub-quadratic time. Bioinformatics 2022, 38, 1532–1541. [Google Scholar] [CrossRef]
- McDonald, D.; Birmingham, A.; Knight, R. Context and the human microbiome. Microbiome 2015, 3, 52. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662. [Google Scholar] [CrossRef]
- Nayfach, S.; Shi, Z.J.; Seshadri, R.; Pollard, K.S.; Kyrpides, N.C. New insights from uncultivated genomes of the global human gut microbiome. Nature 2019, 568, 505–510. [Google Scholar] [CrossRef]
- Mirarab, S.; Nguyen, N.; Warnow, T. SEPP: SATé-Enabled Phylogenetic Placement. In Proceedings of the Pacific Symposium on Biocomputing, Waimea, HI, USA, 3–7 January 2012; World Scientific: Singapore, 2012; pp. 247–258. [Google Scholar] [CrossRef]
- Fitch, W.M.; Margoliash, E. Construction of Phylogenetic Trees. Science 1967, 155, 279–284. [Google Scholar] [CrossRef]
- Felsenstein, J. Inferring Phylogenies; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Desper, R.; Gascuel, O. Fast and Accurate Phylogeny Reconstruction Algorithms Based on the Minimum-Evolution Principle. J. Comput. Biol. 2002, 9, 687–705. [Google Scholar] [CrossRef]
- Erdos, P.; Steel, M.; Szekely, L.; Warnow, T. A few logs suffice to build (almost) all trees: Part II. Theor. Comput. Sci. 1999, 221, 77–118. [Google Scholar] [CrossRef]
- Huson, D.H.; Nettles, S.M.; Warnow, T.J. Disk-covering, a fast-converging method for phylogenetic tree reconstruction. J. Comput. Biol. 1999, 6, 369–386. [Google Scholar] [CrossRef]
- Warnow, T.; Moret, B.M.E.; John, K.S. Absolute convergence: True trees from short sequences. In Proceedings of the Annual ACM-SIAM Symposium on Discrete Algorithms, Washington, DC, USA, 7–9 January 2001. [Google Scholar]
- Roshan, U.; Moret, B.; Warnow, T.; Williams, T. Rec-I-DCM3: A fast algorithmic technique for reconstructing large phylogenetic trees. In Proceedings of the 2004 IEEE Computational Systems Bioinformatics Conference, Washington, DC, USA, 16–19 August 2004; pp. 94–105. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Efron, B. Bootstrap Methods: Another Look at the Jackknife. Ann. Stat. 1979, 7, 1–26. [Google Scholar] [CrossRef]
- Singh, K. On the asymptotic accuracy of Efron’s bootstrap. Ann. Stat. 1981, 9, 1187–1195. [Google Scholar] [CrossRef]
- Susko, E. Bootstrap support is not first-order correct. Syst. Biol. 2009, 58, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Hillis, D.M.; Bull, J.J. An Empirical Test of Bootstrapping as a Method for Assessing Confidence in Phylogenetic Analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Felsenstein, J.; Kishino, H. Is there something wrong with the bootstrap on phylogenies? A reply to Hillis and Bull. Syst. Biol. 1993, 42, 193–200. [Google Scholar] [CrossRef]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef]
- Anisimova, M.; Gascuel, O.; Sullivan, J. Approximate Likelihood-Ratio Test for Branches: A Fast, Accurate, and Powerful Alternative. Syst. Biol. 2006, 55, 539–552. [Google Scholar] [CrossRef]
- Sayyari, E.; Mirarab, S. Fast Coalescent-Based Computation of Local Branch Support from Quartet Frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef]
- Guénoche, A.; Garreta, H. Can We Have Confidence in a Tree Representation? In Proceedings of the Computational Biology, Montpellier, France, 3–5 May 2000; Gascuel, O., Sagot, M.F., Eds.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 45–56. [Google Scholar]
- Cox, D.R. Further Results on Tests of Separate Families of Hypotheses. J. R. Stat. Soc. Ser. B Methodol. 1962, 24, 406–424. [Google Scholar] [CrossRef]
- Goldman, N.; Anderson, J.P.; Rodrigo, A.G. Likelihood-based tests of topologies in phylogenetics. Syst. Biol. 2000, 49, 652–670. [Google Scholar] [CrossRef]
- Rachtman, E.; Sarmashghi, S.; Bafna, V.; Mirarab, S. Uncertainty Quantification Using Subsampling for Assembly-Free Estimates of Genomic Distance and Phylogenetic Relationships. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Politis, D.N.; Romano, J.P.; Wolf, M. Subsampling; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mamm. Protein Metab. 1969, 3, 21–132. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; Hollich, V. Scoredist: A simple and robust protein sequence distance estimator. BMC Bioinform. 2005, 6, 108. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree-2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Guo, S.; Wang, L.S.; Kim, J. Large-scale simulation of RNA macroevolution by an energy-dependent fitness model. arXiv 2009, arXiv:0912.2326. [Google Scholar]
- Zhu, Q.; Mai, U.; Pfeiffer, W.; Janssen, S.; Asnicar, F.; Sanders, J.G.; Belda-Ferre, P.; Al-Ghalith, G.A.; Kopylova, E.; McDonald, D.; et al. WoL: Reference Phylogeny for Microbes (Data Pre-Release). 2019. Available online: https://biocore.github.io/wol/ (accessed on 1 June 2022).
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef]
- Zhu, Q.; Mai, U.; Pfeiffer, W.; Janssen, S.; Asnicar, F.; Sanders, J.G.; Belda-Ferre, P.; Al-Ghalith, G.A.; Kopylova, E.; McDonald, D.; et al. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat. Commun. 2019, 10, 5477. [Google Scholar] [CrossRef]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef]
- Roch, S.; Steel, M. Likelihood-based tree reconstruction on a concatenation of aligned sequence data sets can be statistically inconsistent. Theor. Popul. Biol. 2015, 100, 56–62. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Genes | Placement | Fast BS | Slow BS | Poisson 2 | Binomial 2 | Pplacer | EPA-ng |
|---|---|---|---|---|---|---|---|---|
| RNASIM | 1 | 2.64 | 17.83 | 420.15 | 452.00 | 515.00 | 43.90 | 6.00 |
| Fragmentary (RNASIM) | 1 | 2.40 | 18.80 | - | - | - | 25.85 | 5.00 |
| WOL | 10 | 23.60 | 319.78 | 3289.26 | 899.52 | 967.89 | - | - |
| WOL | 25 | 63.00 | 982.27 | 8851.93 | 1641.77 | 1765.17 | - | - |
| WOL | 50 | 149.01 | 2470.07 | 19,091.11 | 2879.44 | 2959.70 | - | - |
| WOL (Nucleotide) | 1 | 2.30 | 48.77 | - | - | - | 50.77 | 1.23 |
| WOL (AA) | 1 | 2.87 | - | 301.65 | - | - | 45.30 | 0.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, N.B.; Balaban, M.; Biswas, A.; Bayzid, M.S.; Mirarab, S. Distance-Based Phylogenetic Placement with Statistical Support. Biology 2022, 11, 1212. https://doi.org/10.3390/biology11081212
Hasan NB, Balaban M, Biswas A, Bayzid MS, Mirarab S. Distance-Based Phylogenetic Placement with Statistical Support. Biology. 2022; 11(8):1212. https://doi.org/10.3390/biology11081212
Chicago/Turabian StyleHasan, Navid Bin, Metin Balaban, Avijit Biswas, Md. Shamsuzzoha Bayzid, and Siavash Mirarab. 2022. "Distance-Based Phylogenetic Placement with Statistical Support" Biology 11, no. 8: 1212. https://doi.org/10.3390/biology11081212
APA StyleHasan, N. B., Balaban, M., Biswas, A., Bayzid, M. S., & Mirarab, S. (2022). Distance-Based Phylogenetic Placement with Statistical Support. Biology, 11(8), 1212. https://doi.org/10.3390/biology11081212