

Protein–Protein Interaction Network Extraction Using Text Mining Methods Adds Insight into Autism Spectrum Disorder



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Text Pre-Processing
2.2. Sentence Classification Model
2.2.1. Word Embedding
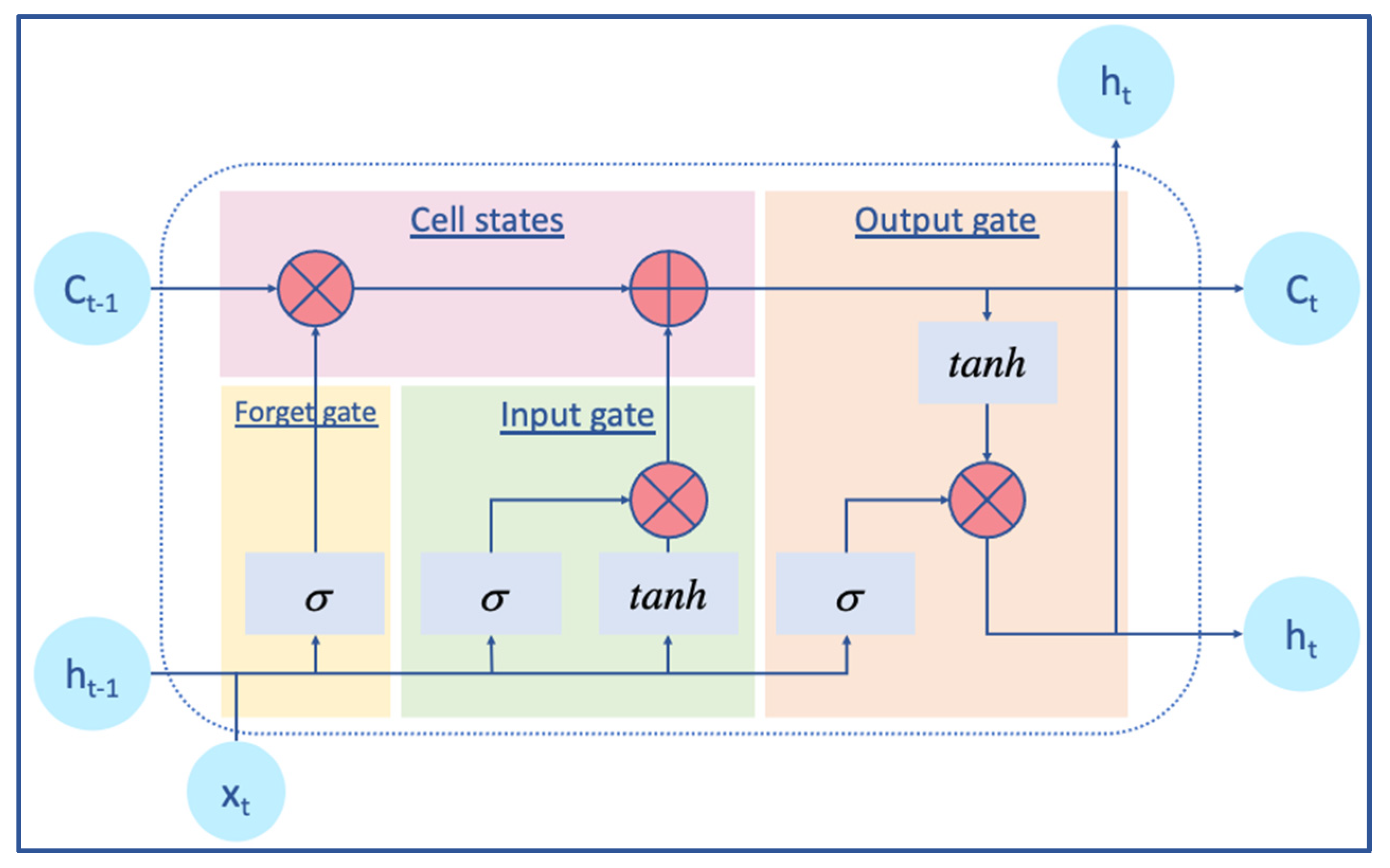
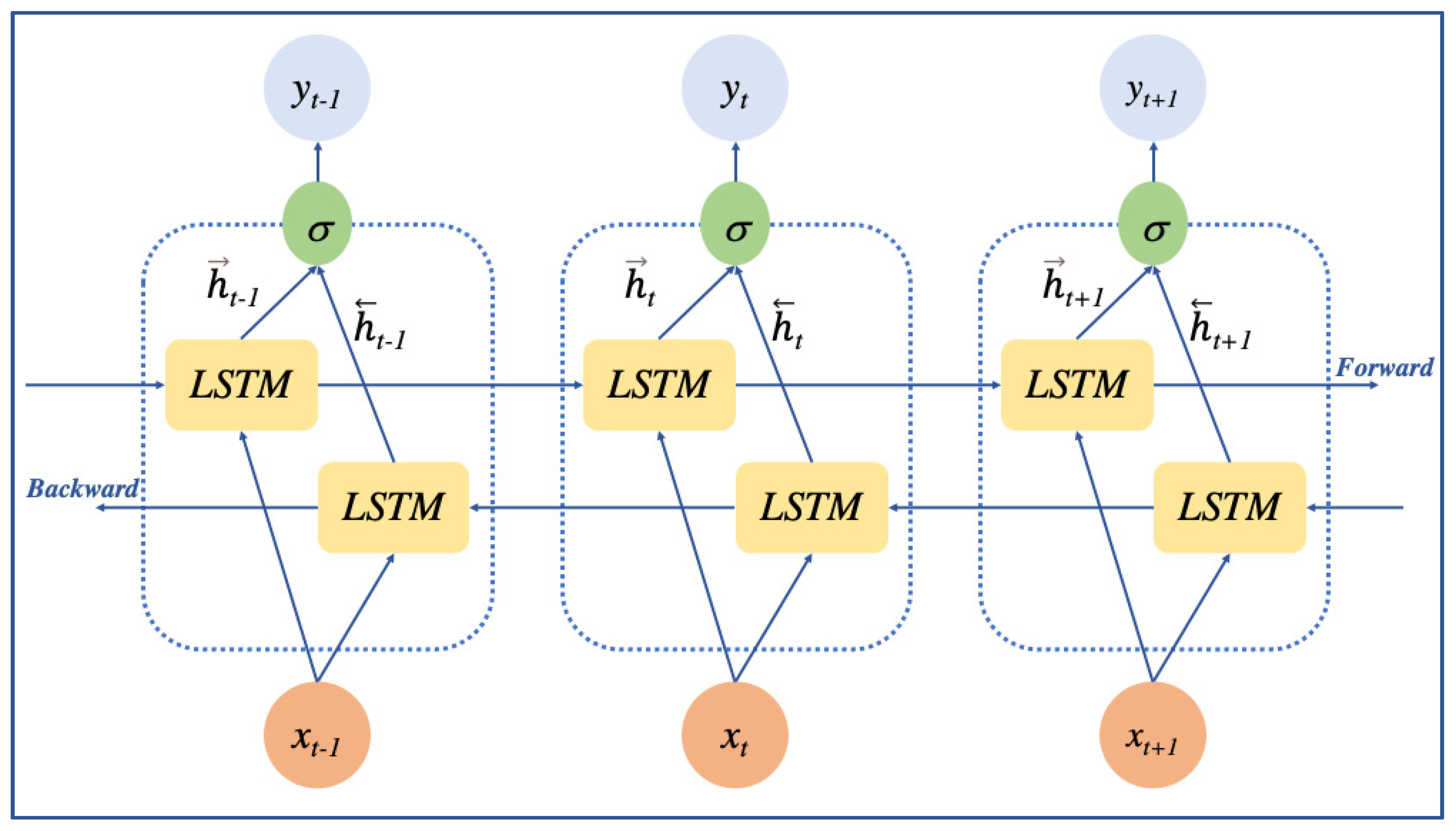
2.2.2. BiLSTM Layer
2.3. Named Entity Recognition Model Using Conditional Random Field
Entity Tagging
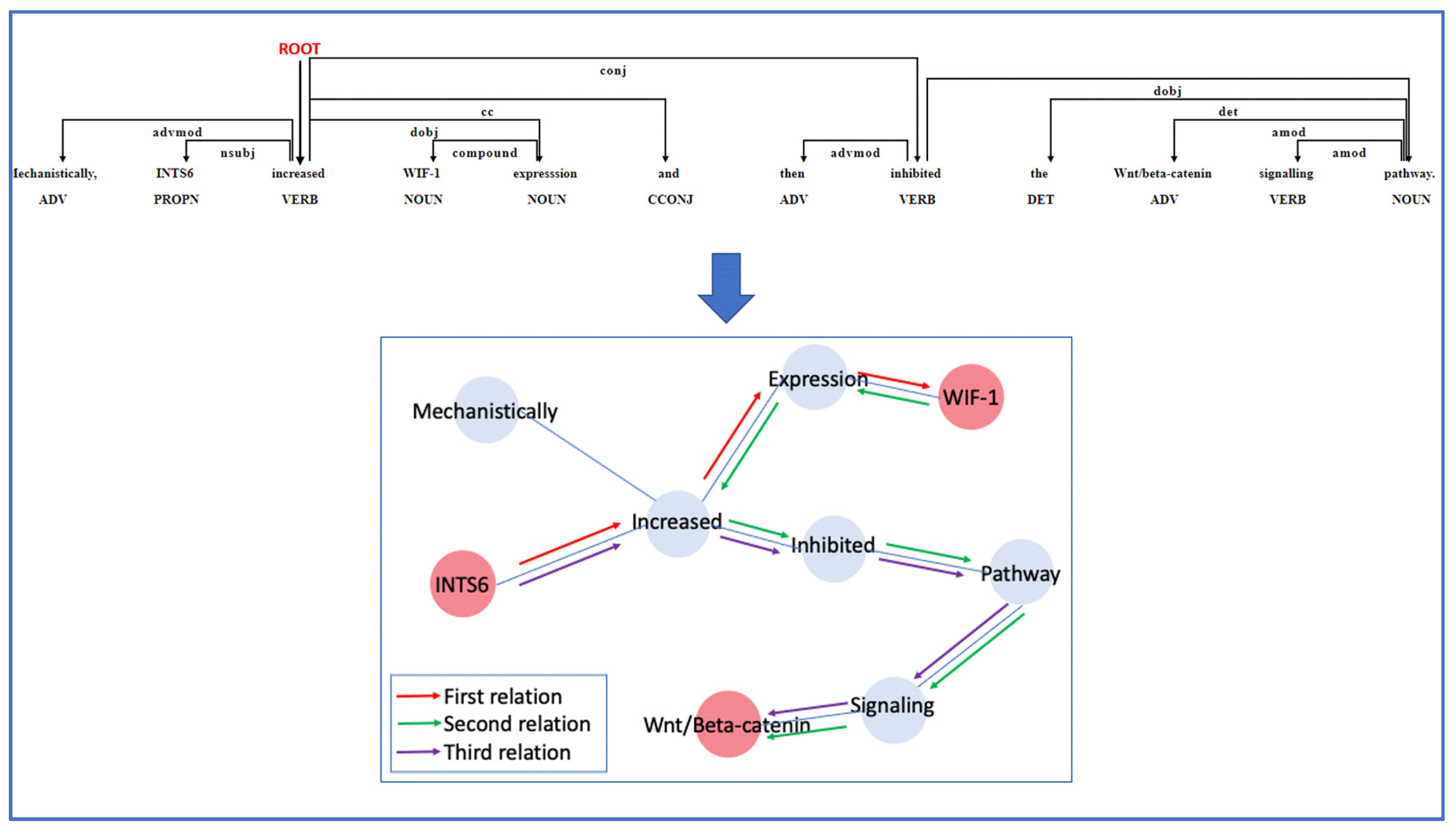
2.4. Relation Extraction
3. Results
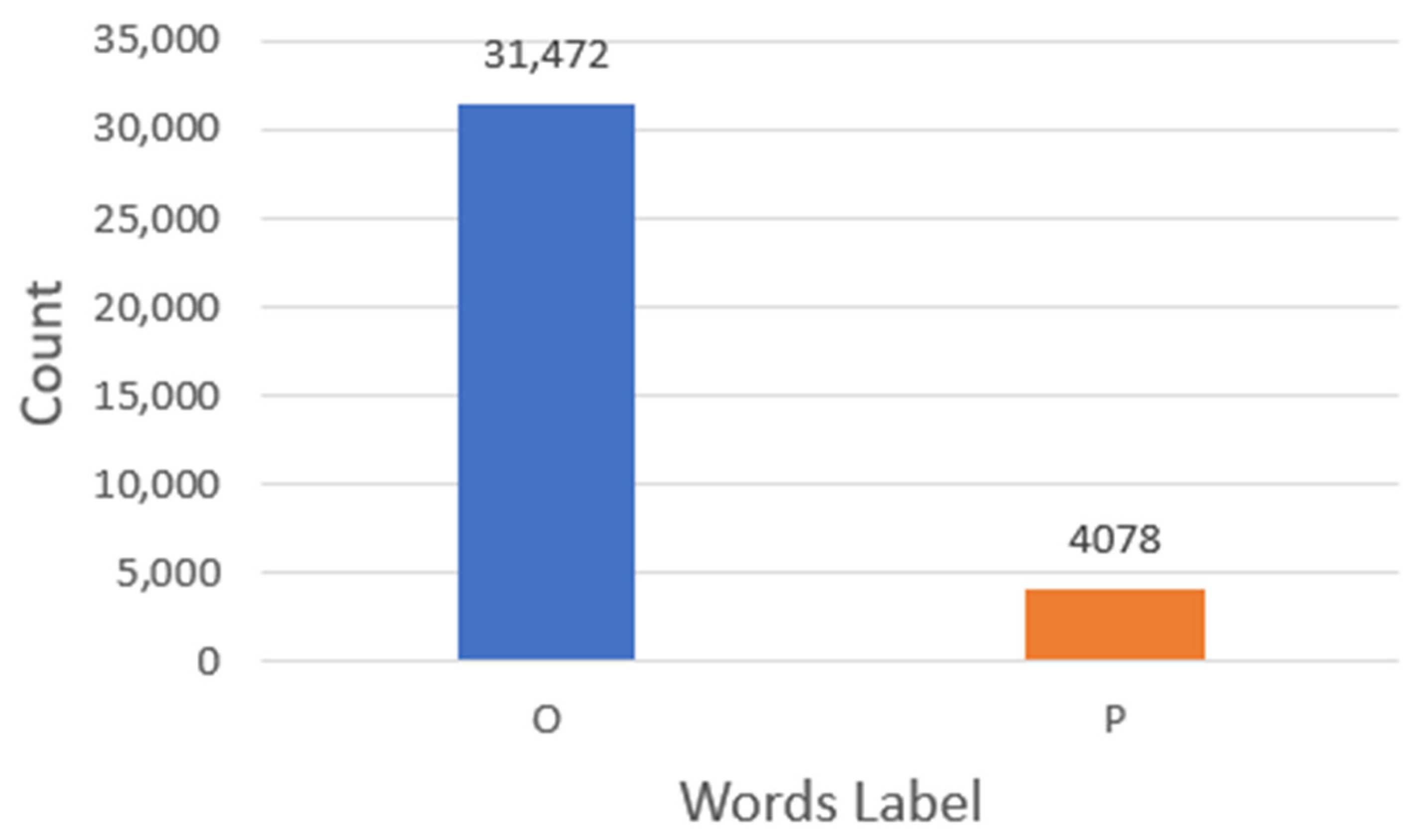
3.1. Data Preparation
3.2. Sentence Classification Models
3.2.1. Word Embedding Initialization
3.2.2. The RNN Layer
3.2.3. Measures of Performance
3.3. Named Entity Recognition Model Initialization
3.4. Relation Extraction Implementation
3.5. Evaluation of Models’ Performance
3.5.1. Sentence Classification Models
3.5.2. NER-CRF Model
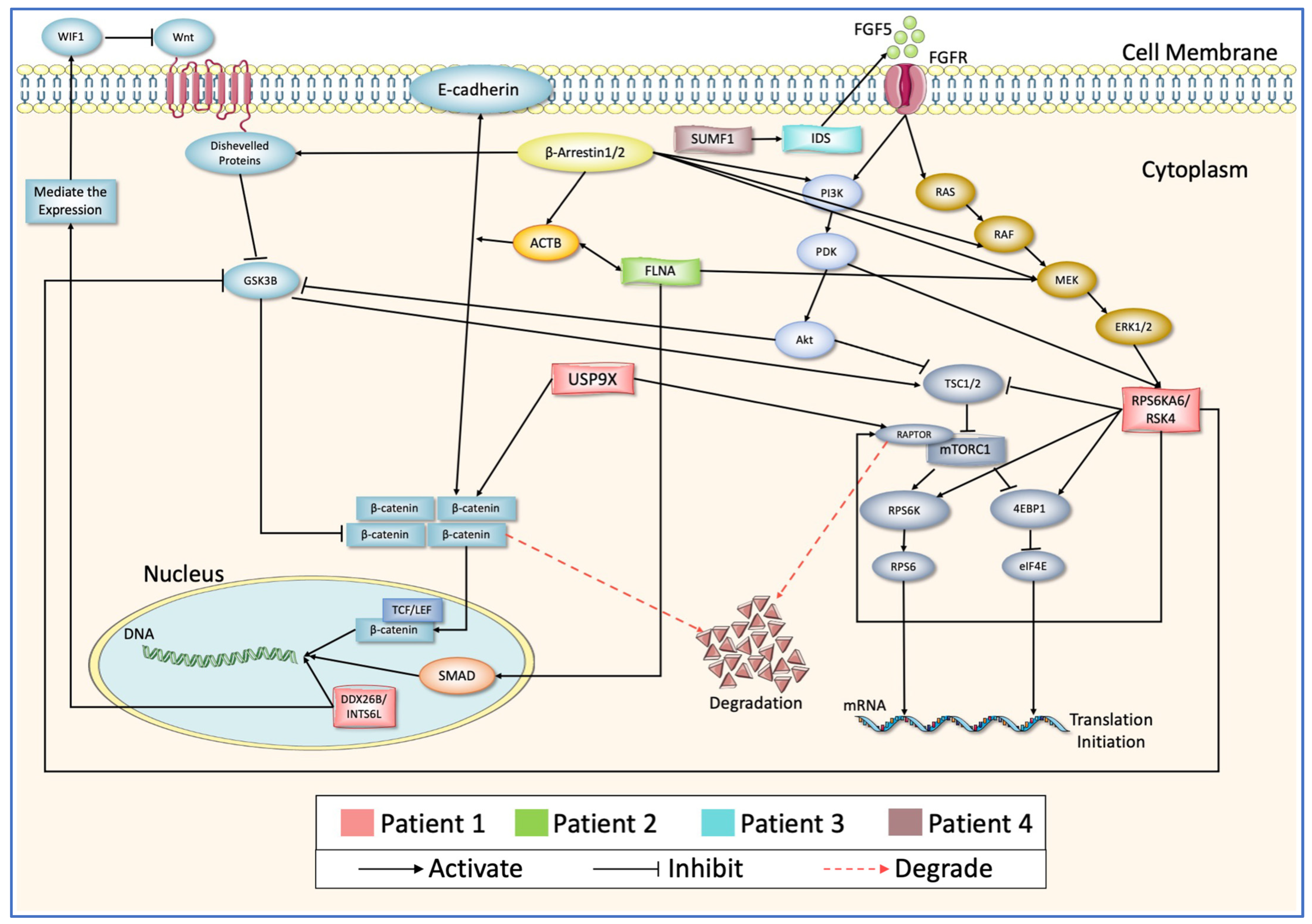
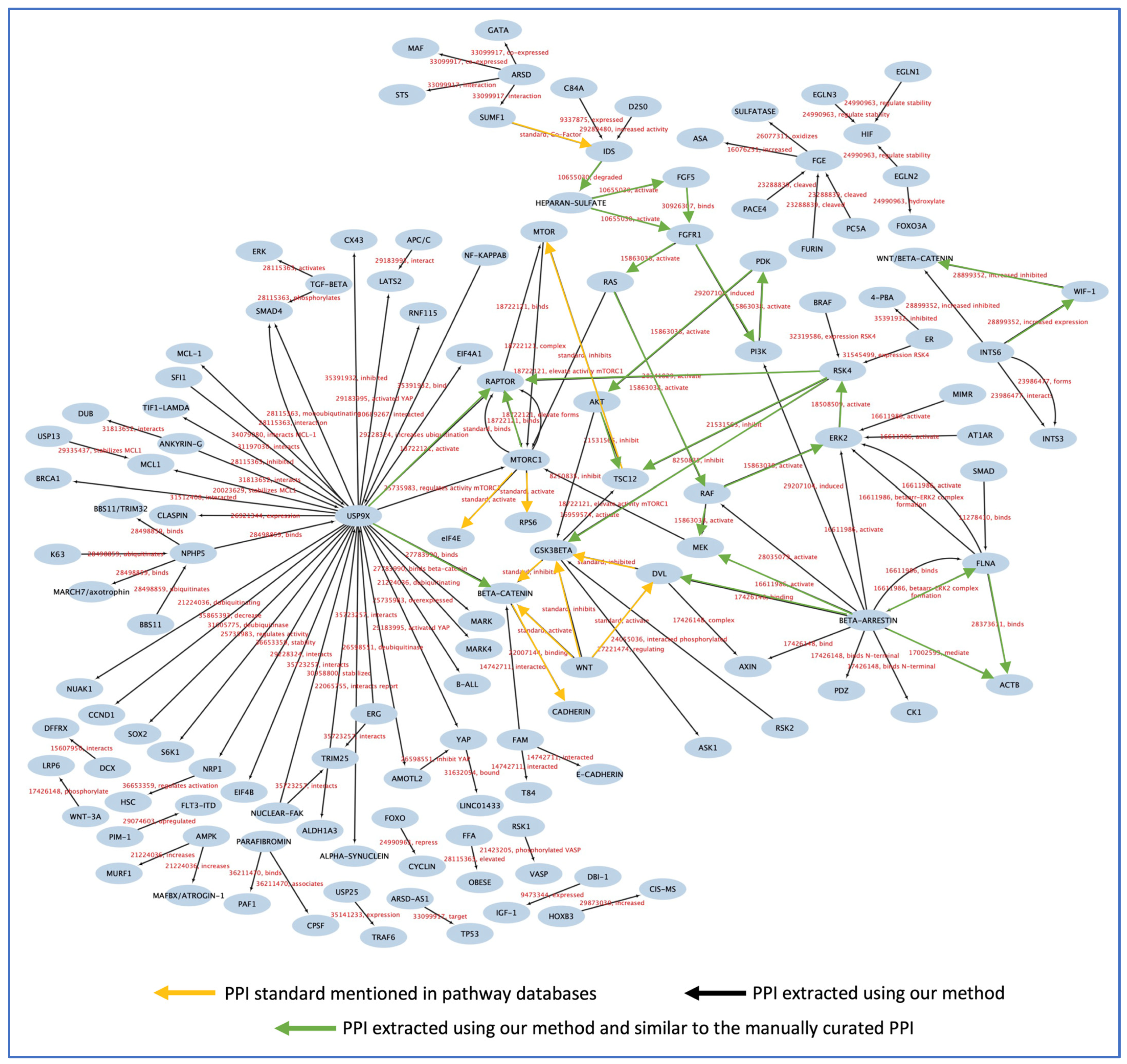
3.6. Testing the Models and PPI Network Creation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Protein function. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Demir, E.; Cary, M.P.; Paley, S.; Fukuda, K.; Lemer, C.; Vastrik, I.; Wu, G.; D’Eustachio, P.; Schaefer, C.; Luciano, J.; et al. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010, 28, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, Ö.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2010, 39, D685–D690. [Google Scholar] [CrossRef] [PubMed]
- Babur, Ö.; Luna, A.; Korkut, A.; Durupinar, F.; Siper, M.C.; Dogrusoz, U.; Jacome, A.S.V.; Peckner, R.; Christianson, K.E.; Jaffe, J.D. Causal interactions from proteomic profiles: Molecular data meet pathway knowledge. Patterns 2021, 2, 100257. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lin, H.; Li, Y. BioPPISVMExtractor: A protein–protein interaction extractor for biomedical literature using SVM and rich feature sets. J. Biomed. Inform. 2010, 43, 88–96. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Airola, A.; Pyysalo, S.; Bjorne, J.; Pahikkala, T.; Ginter, F.; Salakoski, T. All-paths graph kernel for protein-protein interaction extraction with evaluation of cross-corpus learning. BMC Bioinform. 2008, 9 (Suppl. 11), S2. [Google Scholar] [CrossRef]
- Bui, Q.; Katrenko, S.; Sloot, P.M. A hybrid approach to extract protein–protein interactions. Bioinformatics 2011, 27, 259–265. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.; Lee, S.; Lee, K.; Kang, J. High precision rule based PPI extraction and per-pair basis performance evaluation. In Proceedings of the ACM Sixth International Workshop on Data and Text Mining in Biomedical Informatics, Maui, HI, USA, 29 October 2012; pp. 69–76. [Google Scholar]
- Miwa, M.; Sætre, R.; Miyao, Y.; Tsujii, J. A Rich Feature Vector for Protein-Protein Interaction Extraction from Multiple Corpora; Association for Computational Linguistics: Toronto, ON, Canada, 2009; pp. 121–130. [Google Scholar]
- Hsieh, Y.; Chang, Y.; Chang, N.; Hsu, W. Identifying Protein-Protein Interactions in Biomedical Literature Using Recurrent Neural Networks with Long Short-Term Memory; Association for Computational Linguistics: Toronto, ON, Canada, 2017; pp. 240–245. [Google Scholar]
- Hakenberg, J.; Leaman, R.; Vo, N.H.; Jonnalagadda, S.; Sullivan, R.; Miller, C.; Tari, L.; Baral, C.; Gonzalez, G. Efficient extraction of protein-protein interactions from full-text articles. IEEE/ACM Trans. Comput. Biol. Bioinform. 2010, 7, 481–494. [Google Scholar] [CrossRef]
- Hua, L.; Quan, C. A shortest dependency path based convolutional neural network for protein-protein relation extraction. BioMed. Res. Int. 2016, 2016, 8479587. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Munkhdalai, T.; Yu, X.; Ryu, K.H. A novel approach for protein-named entity recognition and protein-protein interaction extraction. Math. Probl. Eng. 2015, 2015, 942435. [Google Scholar] [CrossRef]
- Quan, C.; Luo, Z.; Wang, S. A hybrid deep learning model for protein–protein interactions extraction from biomedical literature. Appl. Sci. 2020, 10, 2690. [Google Scholar] [CrossRef]
- Choi, S. Extraction of protein–protein interactions (PPIs) from the literature by deep convolutional neural networks with various feature embeddings. J. Inf. Sci. 2018, 44, 60–73. [Google Scholar] [CrossRef]
- Peng, Y.; Lu, Z. Deep learning for extracting protein-protein interactions from biomedical literature. arXiv 2017, arXiv:1706.01556. [Google Scholar]
- Gridach, M. Character-level neural network for biomedical named entity recognition. J. Biomed. Inform. 2017, 70, 85–91. [Google Scholar] [CrossRef]
- Zhao, S. Named entity recognition in biomedical texts using an HMM model. In Proceedings of the International Joint Workshop on Natural Language Processing in Biomedicine and Its Applications (NLPBA/BioNLP), Geneva, Switzerland, 28–29 August 2004; pp. 87–90. [Google Scholar]
- Sun, C.; Guan, Y.; Wang, X.; Lin, L. Biomedical named entities recognition using conditional random fields model. In Fuzzy Systems and Knowledge Discovery; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1279–1288. [Google Scholar]
- Sutton, C.; McCallum, A. An introduction to conditional random fields. Found. Trends® Mach. Learn. 2012, 4, 267–373. [Google Scholar] [CrossRef]
- Allot, A.; Peng, Y.; Wei, C.; Lee, K.; Phan, L.; Lu, Z. LitVar: A semantic search engine for linking genomic variant data in PubMed and PMC. Nucleic Acids Res. 2018, 46, W530–W536. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Baumgartner, W.A., Jr.; Randolph, D.A.; Cohen, K.B.; Hunter, L. MutationFinder: A high-performance system for extracting point mutation mentions from text. Bioinformatics 2007, 23, 1862–1865. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Q.; Yang, Z.; Lin, H.; Lu, Z. BioWordVec, improving biomedical word embeddings with subword information and MeSH. Sci. Data 2019, 6, 52. [Google Scholar] [CrossRef]
- Honnibal, M.; Montani, I. spaCy 2: Natural Language Understanding with Bloom Embeddings, Convolutional Neural Networks and Incremental Parsing. Appear 2017, 7, 411–420. [Google Scholar]
- Yin, W.; Kann, K.; Yu, M.; Schütze, H. Comparative study of CNN and RNN for natural language processing. arXiv 2017, arXiv:1702.01923. [Google Scholar]
- Chiu, B.; Crichton, G.; Korhonen, A.; Pyysalo, S. How to train good word embeddings for biomedical NLP. In Proceedings of the 15th Workshop on Biomedical Natural Language Processing, Berlin, Germany, 12 August 2016; pp. 166–174. [Google Scholar]
- Bird, S.; Klein, E.; Loper, E. Natural Language Processing with Python: Analyzing Text with the Natural Language Toolkit; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2009. [Google Scholar]
- Pennington, J.; Socher, R.; Manning, C.D. Glove: Global vectors for word representation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; pp. 1532–1543. [Google Scholar]
- Feng, Y.; Zhang, H.; Hao, W.; Chen, G. Joint extraction of entities and relations using reinforcement learning and deep learning. Comput. Intell. Neurosci. 2017, 2017, 7643065. [Google Scholar] [CrossRef]
- Cai, L.; Zhou, S.; Yan, X.; Yuan, R. A stacked BiLSTM neural network based on coattention mechanism for question answering. Comput. Intell. Neurosci. 2019, 2019, 9543490. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, K.; Jia, S.; Lin, W.; Hou, X.; Liu, B.; Qiu, G. Bidirectional long short-term memory network for vehicle behavior recognition. Remote Sens. 2018, 10, 887. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Gasmi, H.; Laval, J.; Bouras, A. Information extraction of cybersecurity concepts: An LSTM approach. Appl. Sci. 2019, 9, 3945. [Google Scholar] [CrossRef]
- Hagberg, A.; Swart, P.; Chult, S.D. Exploring network structure, dynamics, and function using NetworkX. In Proceedings of the 7th Python in Science Conference, Pasadena, CA, USA, 19–24 August 2008. [Google Scholar]
- Settles, B. ABNER: An open source tool for automatically tagging genes, proteins and other entity names in text. Bioinformatics 2005, 21, 3191–3192. [Google Scholar] [CrossRef]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R.; et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: A trio study from Saudi families. Sci. Rep. 2017, 7, 5679. [Google Scholar] [CrossRef]
- Kapp, L.D.; Abrams, E.W.; Marlow, F.L.; Mullins, M.C. The integrator complex subunit 6 (Ints6) confines the dorsal organizer in vertebrate embryogenesis. PLoS Genet. 2013, 9, e1003822. [Google Scholar] [CrossRef]
- Chen, H.; Shen, H.; Lin, Y.; Mao, Y.; Liu, B.; Xie, L. Small RNA-induced Ints6 gene up-regulation suppresses castration-resistant prostate cancer cells by regulating Β-catenin signaling. Cell Cycle 2018, 17, 1602–1613. [Google Scholar] [CrossRef]
- Lui, K.Y.; Zhao, H.; Qiu, C.; Li, C.; Zhang, Z.; Peng, H.; Fu, R.; Chen, H.; Lu, M. Integrator complex subunit 6 (INTS6) inhibits hepatocellular carcinoma growth by Wnt pathway and serve as a prognostic marker. BMC Cancer 2017, 17, 644. [Google Scholar] [CrossRef] [PubMed]
- Bridges, C.R.; Tan, M.; Premarathne, S.; Nanayakkara, D.; Bellette, B.; Zencak, D.; Domingo, D.; Gecz, J.; Murtaza, M.; Jolly, L.A. USP9X deubiquitylating enzyme maintains RAPTOR protein levels, mTORC1 signalling and proliferation in neural progenitors. Sci. Rep. 2017, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Taya, S.; Yamamoto, T.; Kanai-Azuma, M.; Wood, S.A.; Kaibuchi, K. The deubiquitinating enzyme Fam interacts with and stabilizes β-catenin. Genes Cells 1999, 4, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, S.; Wang, Z.; Yang, C.; Ouyang, W.; Zhou, F.; Zhou, Y.; Xie, C. Deubiquitinase USP9X deubiquitinates β-catenin and promotes high grade glioma cell growth. Oncotarget 2016, 7, 79515. [Google Scholar] [CrossRef]
- Frödin, M.; Jensen, C.J.; Merienne, K.; Gammeltoft, S. A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK. EMBO J. 2000, 19, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef]
- Carrière, A.; Cargnello, M.; Julien, L.; Gao, H.; Bonneil, É.; Thibault, P.; Roux, P.P. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 2004, 101, 13489–13494. [Google Scholar] [CrossRef]
- Roux, P.P.; Shahbazian, D.; Vu, H.; Holz, M.K.; Cohen, M.S.; Taunton, J.; Sonenberg, N.; Blenis, J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J. Biol. Chem. 2007, 282, 14056–14064. [Google Scholar] [CrossRef]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 β by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Esnafoglu, E.; Ayyıldız, S.N. Decreased levels of serum fibroblast growth factor-2 in children with autism spectrum disorder. Psychiatry Res. 2017, 257, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Haub, O.; Drucker, B.; Goldfarb, M. Expression of the murine fibroblast growth factor 5 gene in the adult central nervous system. Proc. Natl. Acad. Sci. USA 1990, 87, 8022–8026. [Google Scholar] [CrossRef] [PubMed]
- Reuss, B.; von Bohlen und Halbach, O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003, 313, 139–157. [Google Scholar] [CrossRef]
- Modarres, H.P.; Mofrad, M.R. Filamin: A structural and functional biomolecule with important roles in cell biology, signaling and mechanics. Mol. Cell. Biomech. 2014, 11, 39. [Google Scholar]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef]
- Sasaki, A.; Masuda, Y.; Ohta, Y.; Ikeda, K.; Watanabe, K. Filamin associates with Smads and regulates transforming growth factor-β signaling. J. Biol. Chem. 2001, 276, 17871–17877. [Google Scholar] [CrossRef]
- Savoy, R.M.; Ghosh, P.M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer 2013, 20, R341–R356. [Google Scholar] [CrossRef]
- Scott, M.G.; Pierotti, V.; Storez, H.; Lindberg, E.; Thuret, A.; Muntaner, O.; Labbé-Jullié, C.; Pitcher, J.A.; Marullo, S. Cooperative regulation of extracellular signal-regulated kinase activation and cell shape change by filamin A and β-arrestins. Mol. Cell. Biol. 2006, 26, 3432–3445. [Google Scholar] [CrossRef]
- Clarke, L.A. The mucopolysaccharidoses: A success of molecular medicine. Expert Rev. Mol. Med. 2008, 10, e1. [Google Scholar] [CrossRef]
- Ornitz, D.M. FGFs, heparan sulfate and FGFRs: Complex interactions essential for development. Bioessays 2000, 22, 108–112. [Google Scholar] [CrossRef]
- Fraldi, A.; Biffi, A.; Lombardi, A.; Visigalli, I.; Pepe, S.; Settembre, C.; Nusco, E.; Auricchio, A.; Naldini, L.; Ballabio, A. SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem. J. 2007, 403, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Annunziata, I.; Roma, G.; Ballabio, A. Sulfatases and sulfatase modifying factors: An exclusive and promiscuous relationship. Hum. Mol. Genet. 2005, 14, 3203–3217. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Combination of AIMed/BioInfer |
|---|---|
| Maximum of sentences length | 120 |
| BiLSTM units | 100 |
| Hidden BiLSTM units | 32 |
| Dropout rate | 0.5 |
| Recurrent dropout rate | 0.2 |
| Optimization algorithm | Adam |
| Activation function | Softmax |
| Learning rate | 1 × 10−4 |
| Epochs | 40 |
| Batch size | 128 |
| Parameter | Combination of AIMed/BioInfer |
|---|---|
| Number of words | 35,550 |
| Algorithm | lbfgs |
| C1 | 0.1 |
| C2 | 0.1 |
| Maximum iterations | 100 |
| All possible transitions | False |
| Method | Positive Class (1) F1 Score | Negative Class (0) F1 Score | Cumulative F1 Score |
|---|---|---|---|
| Bidirectional LSTM + CNN + word embedding (BioNLP) + SDPs embedding [17] | - | - | 0.74 |
| Bidirectional LSTM + word embedding (BioNLP) [13] | - | - | 0.82 |
| CloVe pretrained word embedding + BiLSTM layer | 0.73 | 0.75 | 0.74 |
| BioWordVic pretrained word embedding + BiLSTM layer | 0.76 | 0.77 | 0.77 |
| CloVe pretrained word embedding + 3 hidden layers of BiLSTM | 0.94 | 0.92 | 0.93 |
| BioWordVic pretrained word embedding + 3 hidden layers of BiLSTM | 0.94 | 0.95 | 0.95 |
| Gender | Clinical Demographic Information | Protein Name | Variant Position | Effect of the Variant | ||
|---|---|---|---|---|---|---|
| Mutation Taster | PolyPhen | |||||
| Patient 1 | F | Language delay and regression | DDX26B/INTS6L | p:E435V | DC | PD/0.843 |
| USP9X | p:Y1268C | DC | B/0.007 | |||
| RPS6KA6/RSK4 | p:Q512R | DC | B/0.195 | |||
| Patient 2 | M | NR | FGF5 | p:S84L | DC | D/1.0 |
| FLNA | p:Y2360A | DC | D/0.971 | |||
| Patient 3 | M | Language delay | IDS | p:D175E | DC | PD/0.94 |
| Patient 4 | M | Language delay | SUMF1 | p:Q237R | DC | D/1.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nezamuldeen, L.; Jafri, M.S. Protein–Protein Interaction Network Extraction Using Text Mining Methods Adds Insight into Autism Spectrum Disorder. Biology 2023, 12, 1344. https://doi.org/10.3390/biology12101344
Nezamuldeen L, Jafri MS. Protein–Protein Interaction Network Extraction Using Text Mining Methods Adds Insight into Autism Spectrum Disorder. Biology. 2023; 12(10):1344. https://doi.org/10.3390/biology12101344
Chicago/Turabian StyleNezamuldeen, Leena, and Mohsin Saleet Jafri. 2023. "Protein–Protein Interaction Network Extraction Using Text Mining Methods Adds Insight into Autism Spectrum Disorder" Biology 12, no. 10: 1344. https://doi.org/10.3390/biology12101344
APA StyleNezamuldeen, L., & Jafri, M. S. (2023). Protein–Protein Interaction Network Extraction Using Text Mining Methods Adds Insight into Autism Spectrum Disorder. Biology, 12(10), 1344. https://doi.org/10.3390/biology12101344