
Comparative Analysis of Complete Mitochondrial Genome of Ariosoma meeki (Jordan and Snider, 1900), Revealing Gene Rearrangement and the Phylogenetic Relationships of Anguilliformes
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, and PCR Amplification and Sequencing
2.2. Sequence Analysis and Assembly and Mitochondrial Genome Annotation
2.3. Phylogenetic Analyses
3. Results and Discussion
3.1. Genome Structure and Composition
3.2. PCGs and Codon Usage
3.3. Transfer RNAs, Ribosomal RNAs, and D-Loops
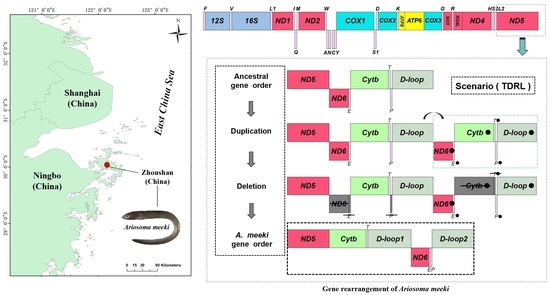
3.4. Gene Rearrangement
3.5. Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bibb, M.J.; Van Etten, R.A.; Wright, C.T.; Walberg, M.W.; Clayton, D.A. Sequence and gene organization of mouse mitochondrial DNA. Cell 1981, 26, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Zhang, L.P.; Yu, D.N.; Storey, K.B.; Zheng, R.Q. Complete mitochondrial genomes of Nanorana taihangnica and N. yunnanensis (Anura: Dicroglossidae) with novel gene arrangements and phylogenetic relationship of Dicroglossidae. BMC Evol. Biol. 2018, 18, 26. [Google Scholar]
- Jie, Y.; Li, H.D.; Zhou, K.Y. Evolution of the mitochondrial genome in snakes: Gene rearrangements and phylogenetic relationships. BMC Genom. 2008, 9, 569. [Google Scholar]
- Gong, L.; Shi, W.; Yang, M.; Li, D.H.; Kong, X.Y. Novel gene arrangement in the mitochondrial genome of Bothus myriaster (Pleuronectiformes: Bothidae): Evidence for the Dimer-Mitogenome and Non-random Loss model. Mitochondrial DNA Part A 2014, 27, 3089–3092. [Google Scholar] [CrossRef]
- Song, X.J.; Tang, W.Q. Complete mitochondrial DNA sequence of Brachysomophis crocodilinus (Anguilliformes: Ophichthidae). Mitochondrial DNA Part B 2017, 2, 187–188. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.J.; Castoe, T.A.; Austin, C.C.; Burbrink, F.T.; Herron, M.D.; McGuire, J.A.; Parkinson, C.L.; Pollock, D.D. Comparative mitochondrial genomics of snakes: Extraordinary substitution rate dynamics and functionality of the duplicate control region. BMC Evol. Biol. 2007, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.S. Ecomorphology of the moray bite: Relationship between dietary extremes and morphological diversity. Physiol. Biochem. Zool. 2009, 82, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Inoue, J.G.; Miya, M.; Miller, M.J.; Sado, T.; Hanel, R.; Hatooka, K.; Aoyama, J.; Minegishi, Y.; Nishida, M.; Tsukamoto, K. Deep-ocean origin of the freshwater eels. Biol. Lett. 2010, 6, 363–366. [Google Scholar] [CrossRef]
- Tang, K.L.; Fielitz, C. Phylogeny of moray eels (Anguilliformes: Muraenidae), with a revised classification of true eels (Teleostei: Elopomorpha: Anguilliformes). Mitochondrial DNA 2013, 24, 55–66. [Google Scholar] [CrossRef]
- Lu, Z.M.; Zhu, K.H.; Jiang, H.; Lu, X.T.; Liu, B.J.; Ye, Y.Y.; Jiang, L.H.; Liu, L.Q.; Gong, L. Complete mitochondrial genome of Ophichthus brevicaudatus reveals novel gene order and phylogenetic relationships of Anguilliformes. Int. J. Biol. Macromol. 2019, 135, 609–618. [Google Scholar] [CrossRef]
- Poulton, J.; Deadman, M.E.; Bindoff, L.; Morten, K.; Land, J.; Brown, G. Families of mtDNA re-arrangements can be detected in patients with mtDNA deletions: Duplications may be a transient intermediate form. Hum. Mol. Genet. 1993, 2, 23–30. [Google Scholar] [CrossRef]
- Ladoukakis, E.D.; Zouros, E. Recombination in animal mitochondrial DNA: Evidence from published sequences. Mol. Biol. Evol. 2001, 18, 2127–2131. [Google Scholar] [CrossRef] [Green Version]
- Christoph, B.; Svenja, S.; Ralph, T. Full mitochondrial genome sequences of two endemic Philippine hornbill species (Aves: Bucerotidae) provide evidence for pervasive mitochondrial DNA recombination. BMC Genom. 2011, 12, 35. [Google Scholar]
- Arndt, A.; Smith, M.J. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol. Biol. Evol. 2018, 8, 1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritz, C.; Brown, T. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Schirtzinger, E.E.; Tavares, E.S.; Gonzales, L.A.; Eberhard, J.R.; Miyaki, C.Y.; Sanchez, J.J.; Hernandez, A.; Mueller, H.; Graves, G.R.; Fleischer, R.C.; et al. Multiple independent origins of mitochondrial control region duplications in the order Psittaciformes. Mol. Phylogenet. Evol. 2012, 64, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, D.S.; Gower, D.J.; Wilkinson, Z.M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol. Biol. Evol. 2006, 23, 227–234. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Boore, J.L.; Brown, W.M. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: Duplication and nonrandom loss. Mol. Biol. Evol. 2002, 19, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, G.H.; Cui, Z.X.; Hui, M.; Liu, Y.; Chan, T.Y.; Song, C.W. The complete mitochondrial genomes of Umalia orientalis and Lyreidus brevifrons: The phylogenetic position of the family Raninidae within Brachyuran crabs. Mar. Genom. 2015, 21, 53–61. [Google Scholar] [CrossRef]
- Mehta, R.S.; Wainwright, P.C. Raptorial jaws in the throat help moray eels swallow large prey. Nature 2007, 449, 79–82. [Google Scholar] [CrossRef]
- Zhang, K.; Zhu, K.H.; Liu, Y.F.; Zhang, H.; Gong, L.; Jiang, L.H.; Liu, L.Q.; Lu, Z.M.; Liu, B.J. Novel gene rearrangement in the mitochondrial genome of Muraenesox cinereus and the phylogenetic relationship of Anguilliformes. Sci. Rep. 2021, 11, 2411. [Google Scholar] [CrossRef] [PubMed]
- Jourda, C.; Santini, S.; Rocher, C.; Le, B.A.; Claverie, J. Mitochondrial genome sequence of the glass sponge Oopsacas minuta. Genome Announcements. 2011, 3, e00823-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wu, P.D.; Zhang, D.Z.; Zhang, H.B.; Tang, B.P.; Liu, Q.N.; Dai, L.S. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 2018, 111, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koichiro, T.; Glen, S.; Sudhir, K. MEGA11: Molecular evolutionary genetics analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Day, J.J. Fishes of the World, 4th Edition. Fish Fish. 2006, 7, 334. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 6, 1550–1552. [Google Scholar] [CrossRef] [Green Version]
- Miya, M.; Takeshima, H.; Endo, H.; Ishiguro, N.B.; Inoue, J.G.; Mukai, T.; Satoh, T.P.; Yamaguchi, M.; Kawaguchi, A.; Mabuchi, K.; et al. Major patterns of higher teleostean phylogenies: A new perspective based on 100 complete mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2003, 26, 121–138. [Google Scholar] [CrossRef]
- Larkin, M.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X v. 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Gerard, T.; Jose, C. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar]
- Xia, X. DAMBE6: New Tools for Microbial Genomics, Phylogenetics, and Molecular Evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.A.; Lopez, J.A.; Satoh, T.P.; Chen, W.J.; Miya, M. Mitochondrial genomic investigation of flatfish monophyly. Gene 2014, 551, 176–182. [Google Scholar] [CrossRef]
- Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar]
- Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [Green Version]
- Sitnikova, T.; Rzhetsky, A.; Nei, M. Interior-branch and bootstrap tests of phylogenetic trees. Mol. Biol. Evol. 1995, 2, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Puts, D.A. Beauty and the beast: Mechanisms of sexual selection in humans. Evol. Hum. Behav. 2010, 31, 157–175. [Google Scholar] [CrossRef]
- Drummond, A.J. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [Green Version]
- Minegishi, Y.; Aoyama, J.; Inoue, J.G.; Miya, M.; Nishida, M.; Tsukamoto, K. Molecular phylogeny and evolution of the freshwater eels genus Anguilla based on the whole mitochondrial genome sequences. Mol. Phylogenet. Evol. 2005, 34, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. Mitogenomic evidence for the monophyly of elopomorph fishes (Teleostei) and the evolutionary origin of the leptocephalus larva. Mol. Phylogenet. Evol. 2004, 32, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. Evolution of the deep-sea gulper eel mitochondrial genomes: Large-scale gene rearrangements originated within the eels. Mol. Biol. Evol. 2003, 20, 1917–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Dong, X.L.; Wang, Z.M.; Miao, X.G.; Wang, S.Y.; Kong, X.Y. Complete mitogenome sequences of four flatfishes (Pleuronectiformes) reveal a novel gene arrangement of L-strand coding genes. BMC Evol. Biol. 2013, 13, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Lu, Z.M.; Guo, B.Y.; Ye, Y.Y.; Liu, L.Q. Characterization of the complete mitochondrial genome of the tidewater goby, Eucyclogobius newberryi (Gobiiformes; Gobiidae; Gobionellinae) and its phylogenetic implications. Conserv. Genet. Resour. 2018, 10, 93–97. [Google Scholar] [CrossRef]
- Elmerot, C.; Arnason, U.; Gojobori, T.; Janke, A. The mitochondrial genome of the pufferfish, Fugu rubripes, and ordinal teleostean relationships. Gene 2002, 295, 163–172. [Google Scholar] [CrossRef]
- Ponce, M.; Infante, C.; Jimenez-Canfizano, R.M.; Perez, L.; Manchado, M. Complete mitochondrial genome of the blackspot seabream, Pagellus bogaraveo (Perciformes: Sparidae), with high levels of length heteroplasmy in the WANCY region. Gene 2008, 409, 44–52. [Google Scholar] [CrossRef]
- Lin, J.P.; Tsai, M.H.; Kroh, A.; Trautman, A.; Machado, D.J.; Chang, L.Y.; Reid, R.; Lin, K.T.; Bronstein, O.; Lee, S.J.; et al. The first complete mitochondrial genome of the sand dollar Sinaechinocyamus mai (Echinoidea: Clypeasteroida). Genomics 2020, 112, 1686–1693. [Google Scholar] [CrossRef]
- Xu, T.J.; Cheng, Y.Z.; Sun, Y.N.; Shi, G.; Wang, R.X. The complete mitochondrial genome of bighead croaker, Collichthys niveatus (Perciformes, Sciaenidae): Structure of control region and phylogenetic considerations. Mol. Biol. Rep. 2011, 38, 4673–4685. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Prabhu, V.R.; Singha, H.S.; Kumar, R.G.; Gopalakrishnan, A.; Nagarajan, M. Characterization of the complete mitochondrial genome of Barilius malabaricus and its phylogenetic implications. Genomics 2020, 112, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Wang, J.; He, S.P.; Mayden, R.L. The complete mitochondrial genome of the Chinese hook snout carp Opsariichthys bidens (Actinopterygii: Cypriniformes) and an alternative pattern of mitogenomic evolution in vertebrate. Gene 2007, 399, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.P.; Pape, T.; Elgar, M.A.; Gao, Y.Y.; Zhang, D. Evolutionary history of stomach bot flies in the light of mitogenomics. Syst. Entomol. 2019, 44, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Dutta, C.; Paul, S. Microbial lifestyle and genome signatures. Curr. Genom. 2012, 13, 153–162. [Google Scholar]
- Yan, L.P.; Xu, W.T.; Zhang, D.; Li, J.Q. Comparative analysis of the mitochondrial genomes of flesh flies and their evolutionary implication. Int. J. Biol. Macromol. 2021, 174, 385–391. [Google Scholar] [CrossRef]
- Gong, L.; Liu, B.J.; Lu, Z.M.; Liu, L.Q. Characterization of the complete mitochondrial genome of Wuhaniligobius polylepis (Gobiiformes: Gobiidae) and phylogenetic studies of Gobiiformes. Mitochondrial DNA Part B 2018, 3, 1117–1119. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Miao, X.G.; Kong, X.Y. A novel model of double replications and random loss accounts for rearrangements in the Mitogenome of Samariscus latus (Teleostei: Pleuronectiformes). BMC Genom. 2014, 15, 352. [Google Scholar] [CrossRef] [Green Version]
- Eberhard, J.R.; Wright, T.F. Rearrangement and evolution of mitochondrial genomes in parrots. Mol. Phylogenet. Evol. 2015, 94, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Gong, L.; Wang, S.Y.; Miao, X.G.; Kong, X.Y. Tandem duplication and random loss for mitogenome rearrangement in Symphurus (Teleost: Pleuronectiformes). BMC Genom. 2015, 16, 355. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Jiang, H.; Zhu, K.H.; Lu, X.T.; Liu, L.Q.; Liu, B.J.; Jiang, L.H.; Ye, Y.Y.; Lu, Z.M. Large-scale mitochondrial gene rearrangements in the hermit crab Pagurus nigrofascia and phylogenetic analysis of the Anomura. Gene 2019, 695, 75–83. [Google Scholar] [CrossRef]
- Chen, J.N.; Lopez, J.A.; Lavoue, S.; Miya, M.; Chen, W.J. Phylogeny of the Elopomorpha (Teleostei): Evidence from six nuclear and mitochondrial markers. Mol. Phylogenet. Evol. 2014, 70, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Forey, P.L.; Littlewood, D.T.; Ritchie, P.; Meyer, A. Interrelationships of Elopomorph Fishes. Interrelationships of Fishes. 1996, 175–191. [Google Scholar]
- Inoue, J.G.; Miya, M.; Tsukamoto, K.; Nishida, M. Complete mitochondrial DNA sequence of Conger myriaster (Teleostei: Anguilliformes): Novel gene order for vertebrate mitochondrial genomes and the phylogenetic implications for anguilliform families. J. Mol. Evol. 2001, 52, 311. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Family | Size (bp) | Accession No. | References |
|---|---|---|---|---|
| Leptocephalus sp. | Nettastomatidae | 18,037 bp | NC_013615 | [8] |
| Nettastoma parviceps | Nettastomatidae | 17,714 bp | NC_013625 | [8] |
| Facciolella oxyrhyncha | Nettastomatidae | 17,789 bp | NC_013621 | [8] |
| Hoplunnis punctata | Nettastomatidae | 17,828 bp | NC_013623 | [8] |
| Paraconger notialis | Congridae | 17,729 bp | NC_013630 | [8] |
| Heteroconger hassi | Congridae | 17,768 bp | NC_013629 | [8] |
| Conger japonicu | Congridae | 17,778 bp | KR131863 | [8] |
| Ariosoma shiroanago | Congridae | 16,922 bp | NC_013632 | [8] |
| Ariosoma meeki | Congridae | 17,659 bp | OK585090 | This Study |
| Cynoponticus ferox | Muraenesocidae | 17,822 bp | NC_013617 | [8] |
| Muraenesox bagio | Muraenesocidae | 18,247 bp | NC_013614 | [8] |
| Kaupichthys hyoproroides | Chlopsidae | 16,662 bp | NC_013607 | [8] |
| Robinsia catherinae | Chlopsidae | 16,627 bp | NC_013633 | [8] |
| Nessorhamphus ingolfianus | Derichthyidae | 17,782 bp | NC_013608 | [8] |
| Coloconger cadenati | Colocongridae | 17,755 bp | NC_013606 | [8] |
| Moringua microchir * | Moringuidae | 15,858 bp | NC_013602 | [8] |
| Moringua edwardsi | Moringuidae | 16,841 bp | NC_013622 | [8] |
| Ophichthus rotundus | Ophichthidae | 17,785 bp | KY081397 | [5] |
| Pythonichthys microphthalmus | Heterenchelyidae | 17,042 bp | NC_013601 | [8] |
| Serrivomer sector | Serrivomeridae | 16,099 bp | NC_013436 | [8] |
| Stemonidium hypomelas | Serrivomeridae | 16,566 bp | NC_013628 | [8] |
| Anguilla marmorata | Anguillidae | 16,714 bp | NC_006540 | [41] |
| Anguilla interioris | Anguillidae | 16,713 bp | NC_006539 | [41] |
| Anguilla dieffenbachii | Anguillidae | 16,687 bp | NC_006538 | [41] |
| Simenchelys parasitica | Synaphobranchidae | 16,689 bp | NC_013605 | [4] |
| Synaphobranchus kaupii | Synaphobranchidae | 16,166 bp | NC_005805 | [42] |
| Ilyophis brunneus | Synaphobranchidae | 16,682 bp | NC_013634 | [4] |
| Myroconger compressus | Myrocongridae | 16,642 bp | NC_013631 | [4] |
| Scuticaria tigrina | Muraenidae | 16,521 bp | KP874183 | [6] |
| Gymnothorax minor | Muraenidae | 16,575 bp | MK204283 | [42] |
| Rhinomuraena quaesita | Muraenidae | 16,566 bp | NC_013610 | [6] |
| Eurypharynx pelecanoides | Eurypharyngidae | 18,978 bp | AB046473 | [43] |
| Saccopharynx lavenbergi | Saccopharyngidae | 18,495 bp | AB047825 | [43] |
| Mitogenome | Position from/to | Length (bp) | Amino Acid | Start/Stop Codon | Anticodon | Intergenic Region from to (bp) * | Strand # | |
|---|---|---|---|---|---|---|---|---|
| A. meeki | ||||||||
| tRNA-Phe (F) | 1 | 71 | 71 | GAA | 0 | H | ||
| 12S RNA | 72 | 1024 | 953 | 0 | H | |||
| tRNA-Val (V) | 1025 | 1095 | 71 | TAC | 0 | H | ||
| 16S RNA | 1096 | 2794 | 1699 | 0 | H | |||
| tRNA-LeuUUA (L1) | 2795 | 2870 | 76 | TAA | 0 | H | ||
| ND1 | 2871 | 3839 | 969 | 323 | ATG/TAG | 0 | H | |
| tRNA-Ile (I) | 3839 | 3906 | 68 | GAT | −1 | H | ||
| tRNA-Gln (Q) | 3908 | 3978 | 71 | TTG | 1 | L | ||
| tRNA-Met (M) | 3978 | 4046 | 69 | CAT | −1 | H | ||
| ND2 | 4047 | 5093 | 1047 | 349 | ATG/TAG | 0 | H | |
| tRNA-Trp (W) | 5092 | 5163 | 72 | TCA | −2 | H | ||
| tRNA-Ala (A) | 5165 | 5233 | 69 | TGC | 1 | L | ||
| tRNA-Asn (N) | 5235 | 5307 | 73 | GTT | 1 | L | ||
| tRNA-Cys (C) | 5347 | 5410 | 64 | GCA | 39 | L | ||
| tRNA-Tyr (Y) | 5411 | 5481 | 71 | GTA | 0 | L | ||
| COX1 | 5483 | 7090 | 1608 | 536 | GTG/AGA | 1 | H | |
| tRNA-SerUCA (S1) | 7086 | 7156 | 71 | TGA | −5 | L | ||
| tRNA-Asp (D) | 7162 | 7231 | 70 | GTC | 5 | L | ||
| COX2 | 7237 | 7927 | 691 | 230 | ATG/T | 5 | H | |
| tRNA-Lys (K) | 7928 | 8002 | 75 | TTT | 0 | H | ||
| ATP8 | 8004 | 8171 | 168 | 56 | ATG/TAA | 1 | H | |
| ATP6 | 8162 | 8845 | 684 | 228 | ATG/TAA | −10 | H | |
| COX3 | 8845 | 9630 | 786 | 262 | ATG/TAA | −1 | H | |
| tRNA-Gly (G) | 9630 | 9701 | 72 | TCC | −1 | H | ||
| ND3 | 9702 | 10,052 | 351 | 117 | ATG/TAG | 0 | H | |
| tRNA-Arg (R) | 10,051 | 10,120 | 70 | TCG | 0 | H | ||
| ND4L | 10,121 | 10,417 | 297 | 99 | ATG/TAA | 0 | H | |
| ND4 | 10,411 | 11,791 | 1381 | 460 | ATG/T | −7 | H | |
| tRNA-His (H) | 11,792 | 11,862 | 71 | GTG | 0 | H | ||
| tRNA-SerAGC (S2) | 11,863 | 11,933 | 71 | GCT | 0 | H | ||
| tRNA-LeuCUA (L2) | 11,934 | 12,007 | 74 | TAG | 0 | H | ||
| ND5 | 12,008 | 13,846 | 1839 | 613 | ATG/TAG | 0 | H | |
| Cyt b | 13,896 | 15,035 | 1140 | 380 | ATG/TAA | 49 | H | |
| tRNA-Thr(T) | 15,037 | 15,107 | 71 | TGT | 1 | H | ||
| D-loop 1 | 15,108 | 16,076 | 969 | 0 | H | |||
| ND6 | 16,077 | 16,592 | 516 | 172 | ATG/TAG | 0 | L | |
| tRNA-Glu (E) | 16,593 | 16,661 | 69 | TTC | 0 | L | ||
| tRNA-Pro (P) | 16,665 | 16,735 | 71 | TGG | 3 | L | ||
| D-loop 2 | 16,736 | 17,695 | 960 | 0 | H | |||
| A. shiroanago | ||||||||
| tRNA-Phe (F) | 1 | 70 | 70 | GAA | 0 | H | ||
| 12S RNA | 71 | 1032 | 962 | 0 | H | |||
| tRNA-Val (V) | 1033 | 1103 | 71 | TAC | 0 | H | ||
| 16S RNA | 1104 | 2828 | 1725 | 0 | H | |||
| tRNA-LeuUUA (L1) | 2823 | 2899 | 77 | TAA | −6 | H | ||
| ND1 | 2900 | 3862 | 963 | 321 | ATG/TAG | 0 | H | |
| tRNA-Ile (I) | 3862 | 3929 | 68 | GAT | −1 | H | ||
| tRNA-Gln (Q) | 3931 | 4001 | 71 | TTG | 1 | L | ||
| tRNA-Met (M) | 4001 | 4046 | 46 | CAT | −1 | H | ||
| ND2 | 4070 | 5116 | 1047 | 349 | ATG/TAG | 23 | H | |
| tRNA-Trp (W) | 5115 | 5187 | 73 | TCA | −2 | H | ||
| tRNA-Ala (A) | 5189 | 5257 | 69 | TGC | 1 | L | ||
| tRNA-Asn (N) | 5259 | 5331 | 73 | GTT | 1 | L | ||
| tRNA-Cys (C) | 5365 | 5430 | 66 | GCA | 33 | L | ||
| tRNA-Tyr (Y) | 5431 | 5501 | 71 | GTA | 0 | L | ||
| COX1 | 5503 | 7110 | 1608 | 536 | GTG/AGG | 1 | H | |
| tRNA-SerUCA (S1) | 7106 | 7176 | 71 | TGA | −5 | L | ||
| tRNA-Asp (D) | 7182 | 7253 | 72 | GTC | 5 | H | ||
| COX2 | 7258 | 7948 | 691 | 230 | ATG/T | 4 | H | |
| tRNA-Lys (K) | 7949 | 8023 | 75 | TTT | 0 | H | ||
| ATP8 | 8025 | 8192 | 168 | 56 | ATG/TAA | 1 | H | |
| ATP6 | 8183 | 8866 | 684 | 228 | ATG/TAA | −10 | H | |
| COX3 | 8866 | 9651 | 786 | 262 | ATG/TAA | −1 | H | |
| tRNA-Gly (G) | 9651 | 9722 | 72 | TCC | −1 | H | ||
| ND3 | 9723 | 10,073 | 351 | 117 | ATG/TAG | 0 | H | |
| tRNA-Arg (R) | 10,072 | 10,141 | 70 | TCG | −2 | H | ||
| ND4L | 10,142 | 10,438 | 297 | 99 | ATG/TAA | 0 | H | |
| ND4 | 10,432 | 11,811 | 1380 | 460 | ATG/TAA | −7 | H | |
| tRNA-His (H) | 11,813 | 11,881 | 69 | GTG | 1 | H | ||
| tRNA-SerAGC (S2) | 11,882 | 11,952 | 71 | GCT | 0 | H | ||
| tRNA-LeuCUA (L2) | 11,953 | 12,026 | 74 | TAG | 0 | H | ||
| ND5 | 12,027 | 13,877 | 1851 | 617 | ATG/TAA | 0 | H | |
| Cyt b | 13,919 | 15,058 | 1140 | 380 | ATG/TAA | 41 | H | |
| tRNA-Thr (T) | 15,061 | 15,132 | 72 | TGT | 2 | H | ||
| ND6 | 15,287 | 15,802 | 516 | 172 | ATG/TAG | 154 | L | |
| tRNA-Glu (E) | 15,803 | 15,871 | 69 | TTC | 0 | L | ||
| tRNA-Pro (P) | 15,886 | 15,955 | 70 | TGG | 14 | L | ||
| D-loop | 15,956 | 16,922 | 967 | 0 | H | |||
| T | C | A | G | A + T% | AT-Skew | GC-Skew | Length (bp) | |
|---|---|---|---|---|---|---|---|---|
| A. meeki | ||||||||
| Mitogenome | 26.14 | 25.39 | 28.93 | 19.54 | 55.07 | 0.05 | −0.13 | 17,695 |
| ND1 | 28.48 | 28.90 | 20.95 | 21.67 | 49.43 | −0.15 | −0.14 | 969 |
| ND2 | 28.27 | 25.21 | 26.27 | 20.25 | 54.54 | −0.04 | −0.11 | 1047 |
| COX1 | 29.42 | 24.13 | 27.42 | 19.03 | 56.84 | −0.04 | −0.12 | 1608 |
| COX2 | 27.49 | 23.60 | 30.39 | 18.52 | 57.88 | 0.05 | −0.12 | 691 |
| ATP8 | 26.79 | 25.00 | 34.52 | 13.69 | 61.31 | 0.13 | −0.29 | 168 |
| ATP6 | 28.80 | 28.51 | 28.65 | 14.04 | 57.45 | 0.00 | −0.34 | 684 |
| COX3 | 28.50 | 27.36 | 24.04 | 20.10 | 52.54 | −0.09 | −0.15 | 786 |
| ND3 | 29.34 | 29.35 | 22.22 | 19.09 | 51.56 | −0.14 | −0.21 | 351 |
| ND4 | 28.24 | 25.13 | 27.73 | 18.90 | 55.97 | −0.01 | −0.14 | 1381 |
| ND4L | 26.94 | 30.30 | 26.60 | 16.16 | 53.54 | −0.01 | −0.30 | 297 |
| ND5 | 26.97 | 28.11 | 25.72 | 19.12 | 52.69 | −0.02 | −0.19 | 1839 |
| Cytb | 29.47 | 27.81 | 23.95 | 18.77 | 53.42 | −0.10 | −0.19 | 1140 |
| ND6 | 33.33 | 20.93 | 14.53 | 31.21 | 47.86 | −0.39 | 0.20 | 516 |
| tRNA | 27.12 | 20.45 | 29.03 | 23.40 | 56.15 | 0.03 | 0.07 | 1560 |
| rRNA | 20.06 | 22.25 | 33.78 | 23.91 | 53.84 | 0.26 | 0.04 | 2652 |
| D-loop | 25.82 | 22.29 | 36.18 | 15.71 | 62.00 | 0.17 | −0.17 | 1929 |
| A. shiroanago | ||||||||
| Mitogenome | 26.85 | 23.90 | 32.54 | 16.71 | 59.38 | 0.10 | −0.18 | 16,922 |
| ND1 | 29.08 | 24.92 | 28.87 | 17.13 | 57.94 | 0.00 | −0.19 | 963 |
| ND2 | 26.46 | 24.45 | 33.72 | 15.38 | 60.17 | 0.12 | −0.23 | 1047 |
| COX1 | 31.59 | 22.26 | 27.80 | 18.35 | 59.39 | −0.06 | −0.10 | 1608 |
| COX2 | 28.51 | 23.30 | 31.26 | 16.93 | 59.77 | 0.05 | −0.16 | 691 |
| ATP8 | 26.79 | 26.19 | 38.69 | 8.33 | 65.48 | 0.18 | −0.52 | 168 |
| ATP6 | 30.56 | 26.32 | 29.97 | 13.16 | 60.53 | −0.01 | −0.33 | 684 |
| COX3 | 28.75 | 24.94 | 28.24 | 18.07 | 57.00 | −0.01 | −0.16 | 786 |
| ND3 | 31.91 | 25.07 | 27.92 | 15.10 | 59.83 | −0.07 | −0.25 | 351 |
| ND4 | 29.13 | 25.94 | 29.57 | 15.36 | 58.70 | 0.01 | −0.26 | 1380 |
| ND4L | 32.32 | 25.59 | 27.61 | 14.48 | 59.93 | −0.08 | −0.28 | 297 |
| ND5 | 28.31 | 25.07 | 32.36 | 14.26 | 60.67 | 0.07 | −0.27 | 1851 |
| Cytb | 30.44 | 24.82 | 29.65 | 15.09 | 60.09 | −0.01 | −0.24 | 1140 |
| ND6 | 41.67 | 14.73 | 14.92 | 28.68 | 56.59 | −0.47 | 0.32 | 516 |
| tRNA | 28.17 | 20.23 | 29.71 | 21.90 | 57.87 | 0.03 | 0.04 | 1562 |
| rRNA | 20.54 | 22.29 | 36.55 | 20.62 | 57.09 | 0.28 | −0.04 | 2687 |
| D-loop | 26.37 | 17.89 | 40.43 | 15.31 | 66.80 | 0.21 | −0.08 | 967 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Zhu, K.; Yang, Y.; Fang, L.; Liu, Z.; Ye, J.; Jia, C.; Chen, J.; Jiang, H. Comparative Analysis of Complete Mitochondrial Genome of Ariosoma meeki (Jordan and Snider, 1900), Revealing Gene Rearrangement and the Phylogenetic Relationships of Anguilliformes. Biology 2023, 12, 348. https://doi.org/10.3390/biology12030348
Huang Y, Zhu K, Yang Y, Fang L, Liu Z, Ye J, Jia C, Chen J, Jiang H. Comparative Analysis of Complete Mitochondrial Genome of Ariosoma meeki (Jordan and Snider, 1900), Revealing Gene Rearrangement and the Phylogenetic Relationships of Anguilliformes. Biology. 2023; 12(3):348. https://doi.org/10.3390/biology12030348
Chicago/Turabian StyleHuang, Youkun, Kehua Zhu, Yawei Yang, Liancheng Fang, Zhaowen Liu, Jia Ye, Caiyi Jia, Jianbin Chen, and Hui Jiang. 2023. "Comparative Analysis of Complete Mitochondrial Genome of Ariosoma meeki (Jordan and Snider, 1900), Revealing Gene Rearrangement and the Phylogenetic Relationships of Anguilliformes" Biology 12, no. 3: 348. https://doi.org/10.3390/biology12030348