
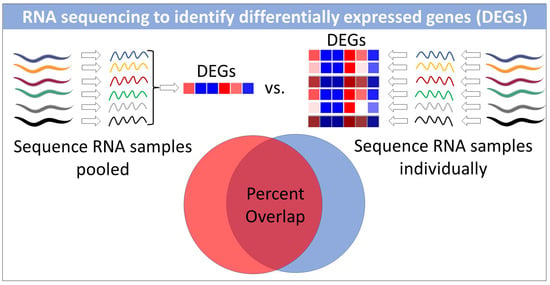
RNA Sequencing of Pooled Samples Effectively Identifies Differentially Expressed Genes



Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Strains
2.2. Isolation of mRNA
2.3. RNA Sequencing and Determination of Differentially Expressed Genes
2.4. Criteria to Identify Differentially Expressed Genes
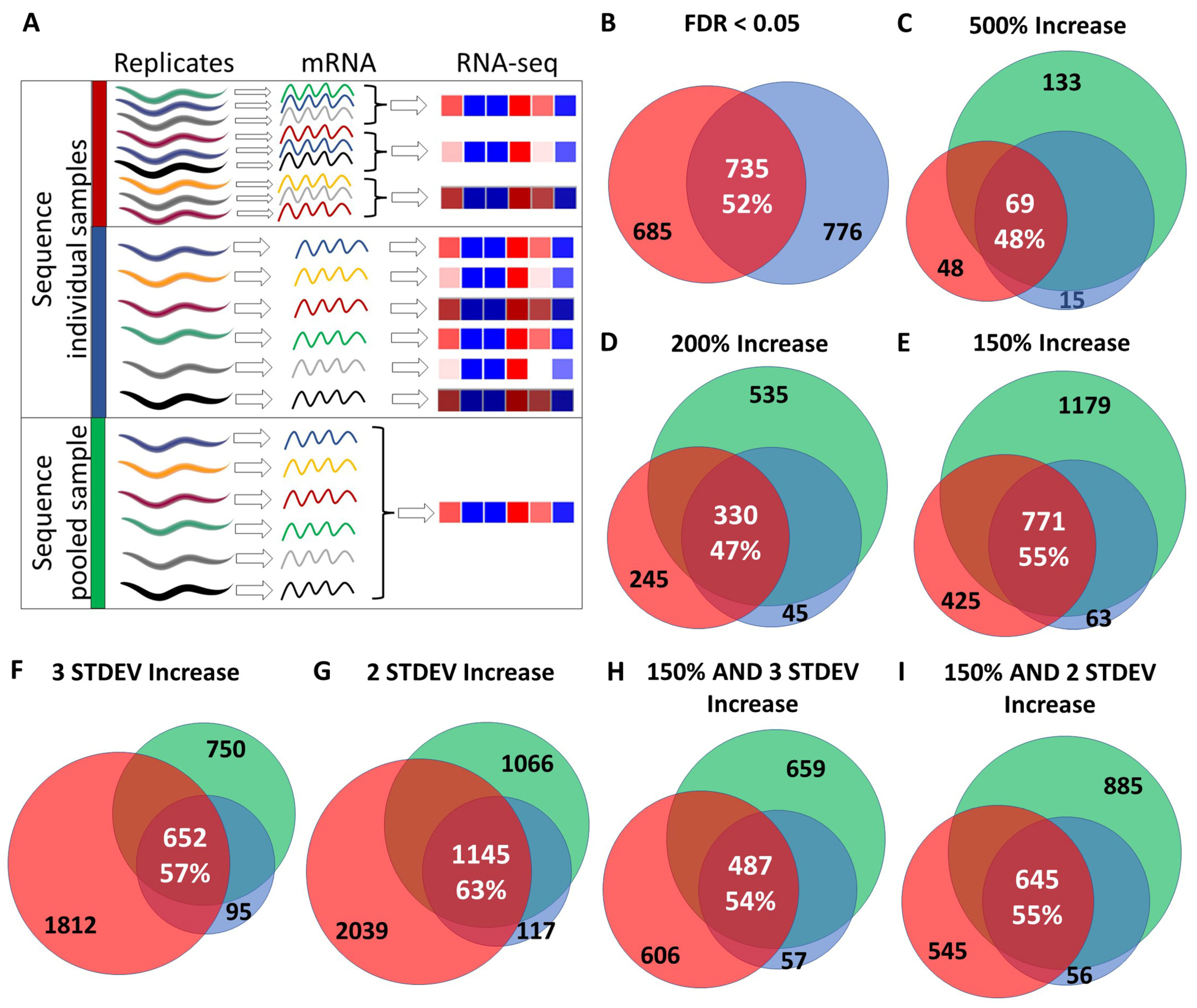
2.5. Comparison of Differentially Expressed Genes
3. Results
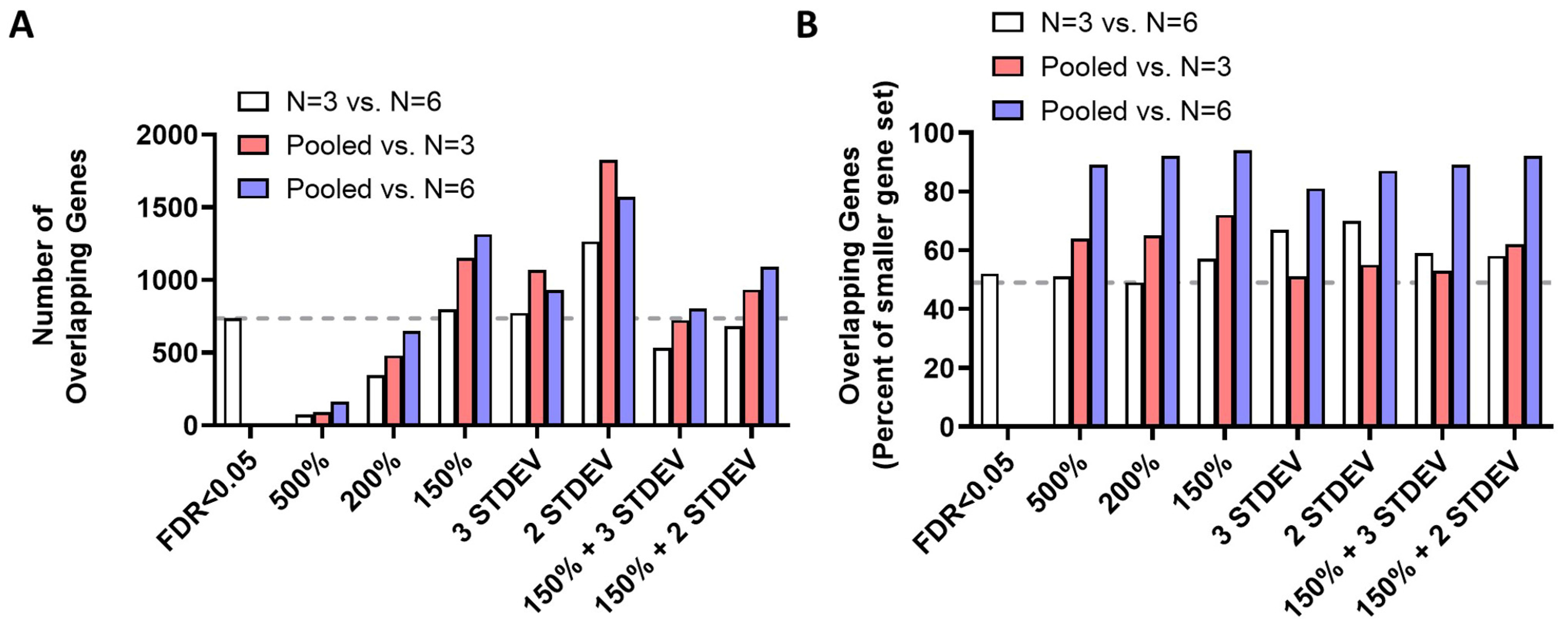
3.1. Reproducibility of RNA Sequencing Results between Experiments
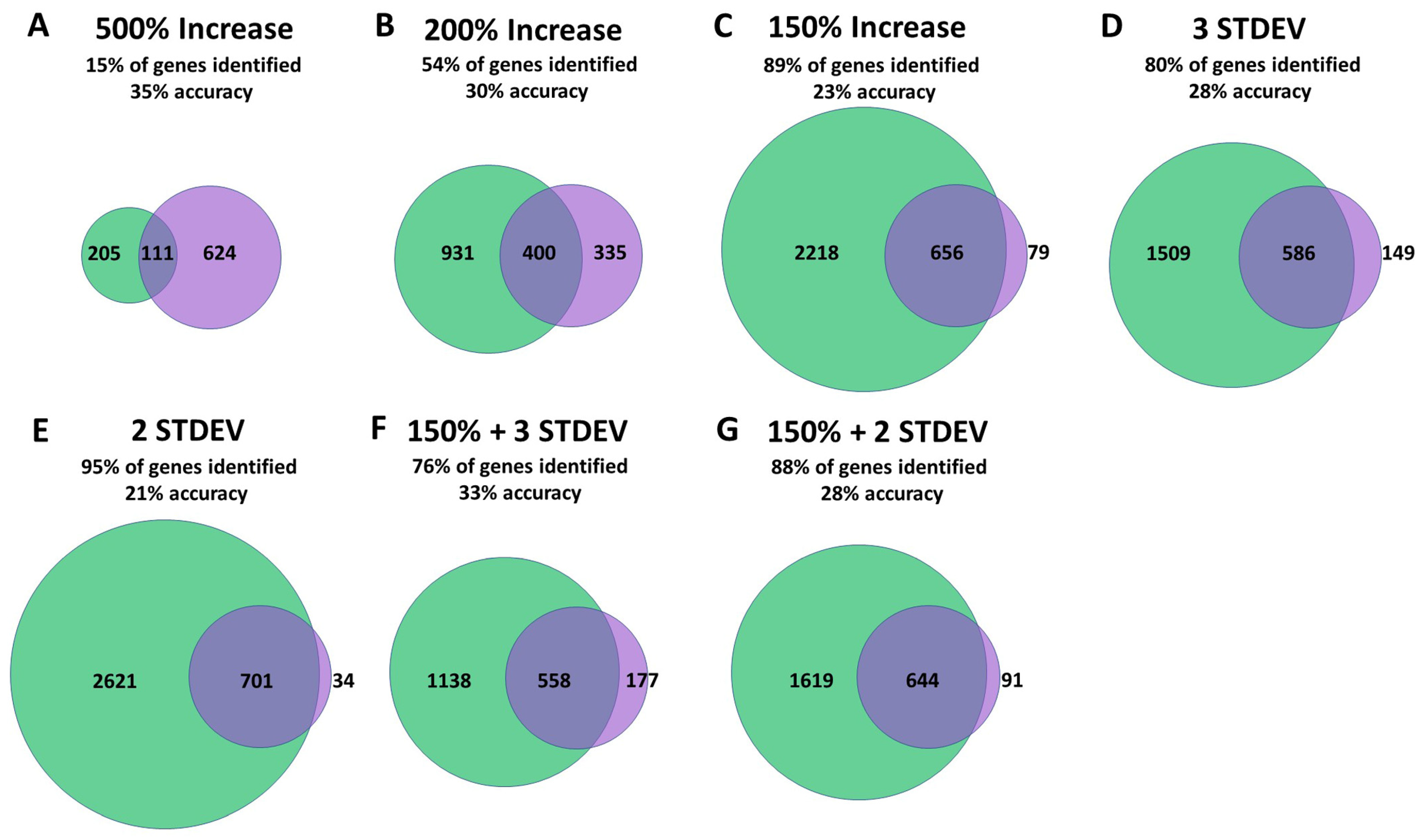
3.2. Differentially Expressed Genes Can Be Identified by Sequencing a Single Pooled RNA Sample
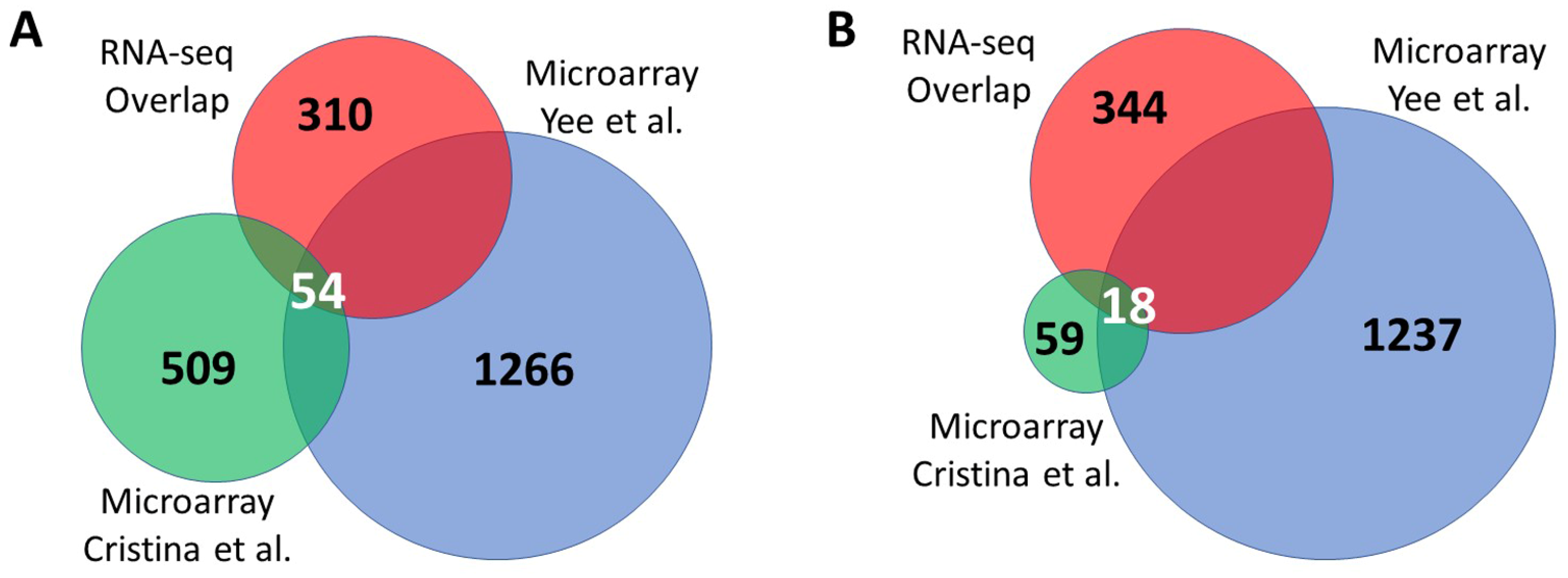
3.3. Overlap between Independent Microarray Experiments and RNA Sequencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Driver, J.A.; Logroscino, G.; Gaziano, J.M.; Kurth, T. Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology 2009, 72, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Senchuk, M.M.; Van Raamsdonk, J.M.; Moore, D.J. Multiple genetic pathways regulating lifespan extension are neuroprotective in a G2019S LRRK2 nematode model of Parkinson’s disease. Neurobiol. Dis. 2021, 151, 105267. [Google Scholar] [CrossRef] [PubMed]
- Soo, S.K.; Rudich, P.D.; Traa, A.; Harris-Gauthier, N.; Shields, H.J.; Van Raamsdonk, J.M. Compounds that extend longevity are protective in neurodegenerative diseases and provide a novel treatment strategy for these devastating disorders. Mech. Ageing Dev. 2020, 190, 111297. [Google Scholar] [CrossRef]
- Morley, J.F.; Brignull, H.R.; Weyers, J.J.; Morimoto, R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 10417–10422. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.A.; Arango, M.; Abderrahmane, S.; Lambert, E.; Tourette, C.; Catoire, H.; Neri, C. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat. Genet. 2005, 37, 349–350. [Google Scholar] [CrossRef]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef]
- Knight, A.L.; Yan, X.; Hamamichi, S.; Ajjuri, R.R.; Mazzulli, J.R.; Zhang, M.W.; Daigle, J.G.; Zhang, S.; Borom, A.R.; Roberts, L.R.; et al. The Glycolytic Enzyme, GPI, Is a Functionally Conserved Modifier of Dopaminergic Neurodegeneration in Parkinson’s Models. Cell Metab. 2014, 20, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Boccitto, M.; Lamitina, T.; Kalb, R.G. Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS ONE 2012, 7, e33494. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.L.; Murphy, C.T.; Kenyon, C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 2003, 300, 1142–1145. [Google Scholar] [CrossRef] [Green Version]
- Lakowski, B.; Hekimi, S. Determination of life-span in Caenorhabditis elegans by four clock genes. Science 1996, 272, 1010–1013. [Google Scholar] [CrossRef]
- Wong, A.; Boutis, P.; Hekimi, S. Mutations in the clk-1 gene of Caenorhabditis elegans affect developmental and behavioral timing. Genetics 1995, 139, 1247–1259. [Google Scholar] [CrossRef]
- Feng, J.; Bussiere, F.; Hekimi, S. Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Hekimi, S. Two modes of mitochondrial dysfunction lead independently to lifespan extension in Caenorhabditis elegans. Aging Cell 2010, 9, 433–447. [Google Scholar] [CrossRef]
- Dues, D.J.; Schaar, C.E.; Johnson, B.K.; Bowman, M.J.; Winn, M.E.; Senchuk, M.M.; Van Raamsdonk, J.M. Uncoupling of oxidative stress resistance and lifespan in long-lived isp-1 mitochondrial mutants in Caenorhabditis elegans. Free Radic. Biol. Med. 2017, 108, 362–373. [Google Scholar] [CrossRef]
- Wu, Z.; Senchuk, M.M.; Dues, D.J.; Johnson, B.K.; Cooper, J.F.; Lew, L.; Machiela, E.; Schaar, C.E.; DeJonge, H.; Blackwell, T.K.; et al. Mitochondrial unfolded protein response transcription factor ATFS-1 promotes longevity in a long-lived mitochondrial mutant through activation of stress response pathways. BMC Biol. 2018, 16, 147. [Google Scholar] [CrossRef] [Green Version]
- Campos, J.C.; Wu, Z.; Rudich, P.D.; Soo, S.K.; Mistry, M.; Ferreira, J.C.; Blackwell, T.K.; Van Raamsdonk, J.M. Mild mitochondrial impairment enhances innate immunity and longevity through ATFS-1 and p38 signaling. EMBO Rep. 2021, 22, e52964. [Google Scholar] [CrossRef] [PubMed]
- Harris-Gauthier, N.; Traa, A.; AlOkda, A.; Moldakozhayev, A.; Anglas, U.; Soo, S.K.; Van Raamsdonk, J.M. Mitochondrial thioredoxin system is required for enhanced stress resistance and extended longevity in long-lived mitochondrial mutants. Redox Biol. 2022, 53, 102335. [Google Scholar] [CrossRef] [PubMed]
- Senchuk, M.M.; Dues, D.J.; Schaar, C.E.; Johnson, B.K.; Madaj, Z.B.; Bowman, M.J.; Winn, M.E.; Van Raamsdonk, J.M. Activation of DAF-16/FOXO by reactive oxygen species contributes to longevity in long-lived mitochondrial mutants in Caenorhabditis elegans. PLoS Genet. 2018, 14, e1007268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soo, S.K.; Traa, A.; Rudich, Z.D.; Moldakozhayev, A.; Mistry, M.; Van Raamsdonk, J.M. Genetic basis of enhanced stress resistance in long-lived mutants highlights key role of innate immunity in determining longevity. Aging Cell 2023, 22, e13740. [Google Scholar] [CrossRef]
- Cristina, D.; Cary, M.; Lunceford, A.; Clarke, C.; Kenyon, C. A regulated response to impaired respiration slows behavioral rates and increases lifespan in Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000450. [Google Scholar] [CrossRef] [Green Version]
- Yee, C.; Yang, W.; Hekimi, S. The Intrinsic Apoptosis Pathway Mediates the Pro-Longevity Response to Mitochondrial ROS in C. elegans. Cell 2014, 157, 897–909. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef]
- McElwee, J.J.; Schuster, E.; Blanc, E.; Thomas, J.H.; Gems, D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 2004, 279, 44533–44543. [Google Scholar] [CrossRef] [Green Version]
- Hwang, A.B.; Ryu, E.A.; Artan, M.; Chang, H.W.; Kabir, M.H.; Nam, H.J.; Lee, D.; Yang, J.S.; Kim, S.; Mair, W.B.; et al. Feedback regulation via AMPK and HIF-1 mediates ROS-dependent longevity in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef] [Green Version]
- Gao, A.W.; Smith, R.L.; van Weeghel, M.; Kamble, R.; Janssens, G.E.; Houtkooper, R.H. Identification of key pathways and metabolic fingerprints of longevity in C. elegans. Exp. Gerontol. 2018, 113, 128–140. [Google Scholar] [CrossRef]
- Greer, E.L.; Maures, T.J.; Hauswirth, A.G.; Green, E.M.; Leeman, D.S.; Maro, G.S.; Han, S.; Banko, M.R.; Gozani, O.; Brunet, A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 2010, 466, 383–387. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Isik, M.; Moroz, N.; Steinbaugh, M.J.; Zhang, P.; Blackwell, T.K. Dietary Restriction Extends Lifespan through Metabolic Regulation of Innate Immunity. Cell Metab. 2019, 29, 1192–1205.e8. [Google Scholar] [CrossRef]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. BioTechniques 2005, 39, 75–85. [Google Scholar] [CrossRef]
- Trevino, V.; Falciani, F.; Barrera-Saldana, H.A. DNA microarrays: A powerful genomic tool for biomedical and clinical research. Mol. Med. 2007, 13, 527–541. [Google Scholar] [CrossRef]
- Dalma-Weiszhausz, D.D.; Warrington, J.; Tanimoto, E.Y.; Miyada, C.G. The affymetrix GeneChip platform: An overview. Methods Enzymol. 2006, 410, 3–28. [Google Scholar] [CrossRef]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.P.; Miaskowski, C.; Dhruva, A.A.; Flowers, E.; Kober, K.M. Mechanisms and Measurement of Changes in Gene Expression. Biol. Res. Nurs. 2018, 20, 369–382. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [Green Version]
- Schulze, A.; Downward, J. Navigating gene expression using microarrays—A technology review. Nat. Cell Biol. 2001, 3, E190–E195. [Google Scholar] [CrossRef]
- Abdullah-Sayani, A.; Bueno-de-Mesquita, J.M.; van de Vijver, M.J. Technology Insight: Tuning into the genetic orchestra using microarrays—Limitations of DNA microarrays in clinical practice. Nat. Clin. Pract. Oncol. 2006, 3, 501–516. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Machiela, E.; Dues, D.J.; Senchuk, M.M.; Van Raamsdonk, J.M. Oxidative stress is increased in C. elegans models of Huntington’s disease but does not contribute to polyglutamine toxicity phenotypes. Neurobiol. Dis. 2016, 96, 1–11. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.N.; Aleksic, J.; Butano, D.; Carr, A.; Contrino, S.; Hu, F.; Lyne, M.; Lyne, R.; Kalderimis, A.; Rutherford, K.; et al. InterMine: A flexible data warehouse system for the integration and analysis of heterogeneous biological data. Bioinformatics 2012, 28, 3163–3165. [Google Scholar] [CrossRef]
- De Las Rivas, J.; Aibar, S.; Roson, B. Gene Expression Analysis and Profiling of Microarrays Data and RNA-Sequencing Data. Compr. Anal. Chem. 2014, 63, 355–384. [Google Scholar]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gong, B.; Bushel, P.R.; Thierry-Mieg, J.; Thierry-Mieg, D.; Xu, J.; Fang, H.; Hong, H.; Shen, J.; Su, Z.; et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol. 2014, 32, 926–932. [Google Scholar] [CrossRef]
- Zhang, W.; Yu, Y.; Hertwig, F.; Thierry-Mieg, J.; Zhang, W.; Thierry-Mieg, D.; Wang, J.; Furlanello, C.; Devanarayan, V.; Cheng, J.; et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015, 16, 133. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.F.; Tycksen, E.D.; Sandell, L.J.; Brophy, R.H. Advantages of RNA-seq compared to RNA microarrays for transcriptome profiling of anterior cruciate ligament tears. J. Orthop. Res. 2018, 36, 484–497. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.S.; Van Vleet, T.R.; Ciurlionis, R.; Buck, W.R.; Mittelstadt, S.W.; Blomme, E.A.G.; Liguori, M.J. Comparison of RNA-Seq and Microarray Gene Expression Platforms for the Toxicogenomic Evaluation of Liver From Short-Term Rat Toxicity Studies. Front. Genet. 2018, 9, 636. [Google Scholar] [CrossRef] [Green Version]
- Black, M.B.; Parks, B.B.; Pluta, L.; Chu, T.M.; Allen, B.C.; Wolfinger, R.D.; Thomas, R.S. Comparison of microarrays and RNA-seq for gene expression analyses of dose-response experiments. Toxicol. Sci. Off. J. Soc. Toxicol. 2014, 137, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.; Liang, F.; Oesterreich, S.; Tseng, G.C. A Joint Bayesian Model for Integrating Microarray and RNA Sequencing Transcriptomic Data. J. Comput. Biol. 2017, 24, 647–662. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, B.; Van Raamsdonk, J.M. RNA Sequencing of Pooled Samples Effectively Identifies Differentially Expressed Genes. Biology 2023, 12, 812. https://doi.org/10.3390/biology12060812
Ko B, Van Raamsdonk JM. RNA Sequencing of Pooled Samples Effectively Identifies Differentially Expressed Genes. Biology. 2023; 12(6):812. https://doi.org/10.3390/biology12060812
Chicago/Turabian StyleKo, Bokang, and Jeremy M. Van Raamsdonk. 2023. "RNA Sequencing of Pooled Samples Effectively Identifies Differentially Expressed Genes" Biology 12, no. 6: 812. https://doi.org/10.3390/biology12060812
APA StyleKo, B., & Van Raamsdonk, J. M. (2023). RNA Sequencing of Pooled Samples Effectively Identifies Differentially Expressed Genes. Biology, 12(6), 812. https://doi.org/10.3390/biology12060812