
Using Zebrafish to Dissect the Interaction of Mycobacteria with the Autophagic Machinery in Macrophages




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Zebrafish Husbandry
2.2. Tail Fin Infection
2.3. Confocal Laser Scanning Microscopy (CLSM)
3. Results
3.1. Tail Fin Infection High-Resolution Live Imaging
3.2. Mm Infection Rapidly Increases the LC3 Levels in Macrophages
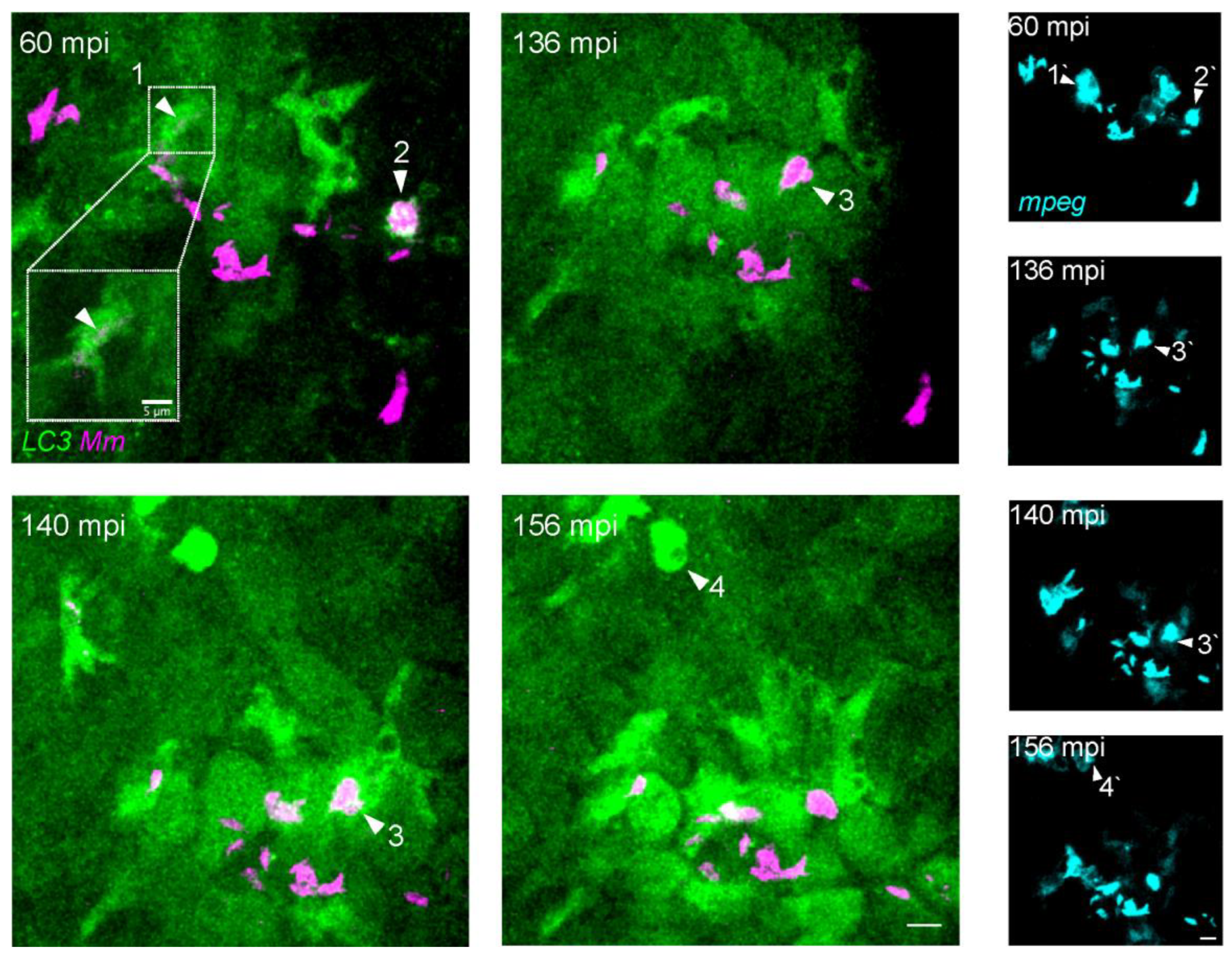
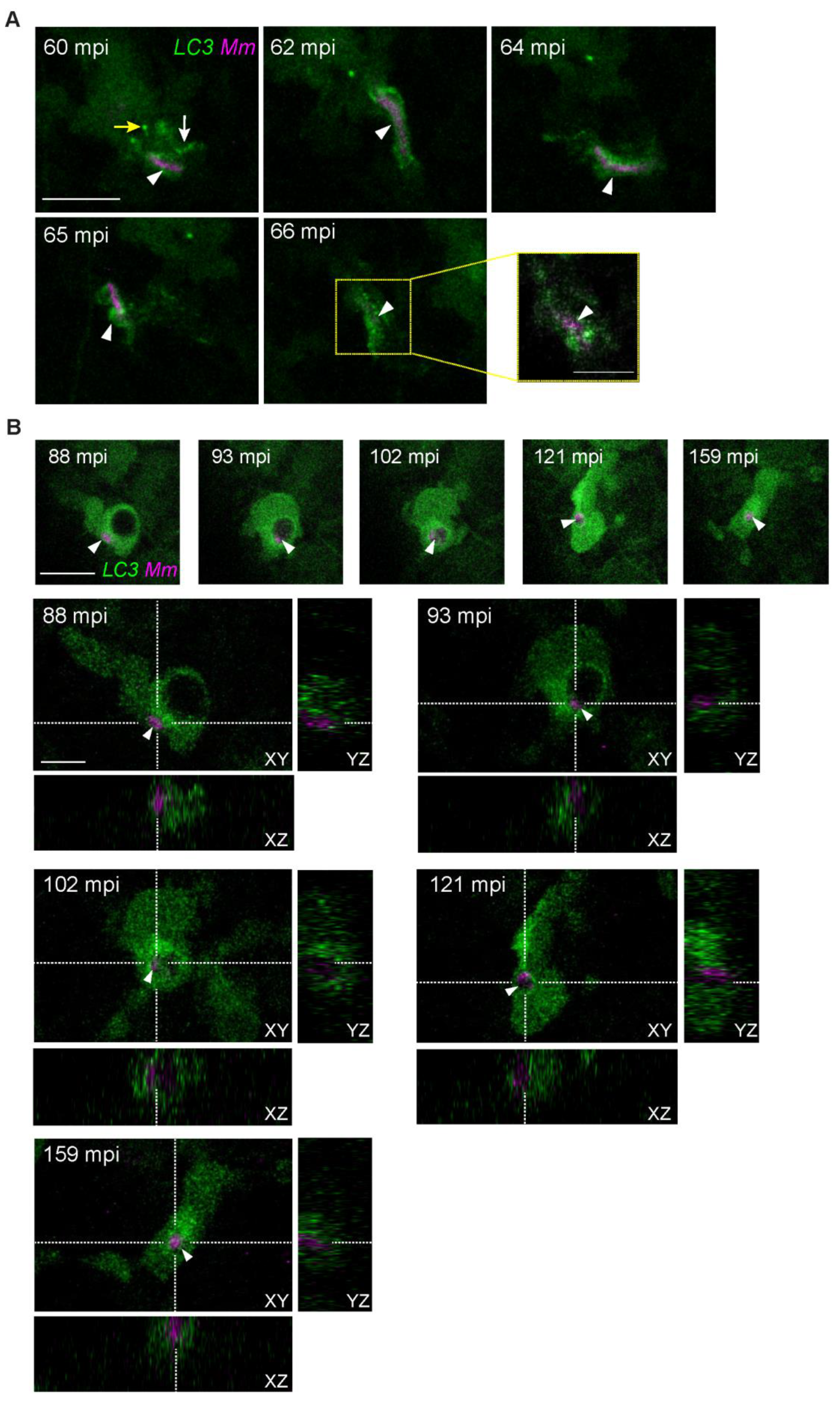
3.3. Intracellular Dynamics of LC3 Association with Mm
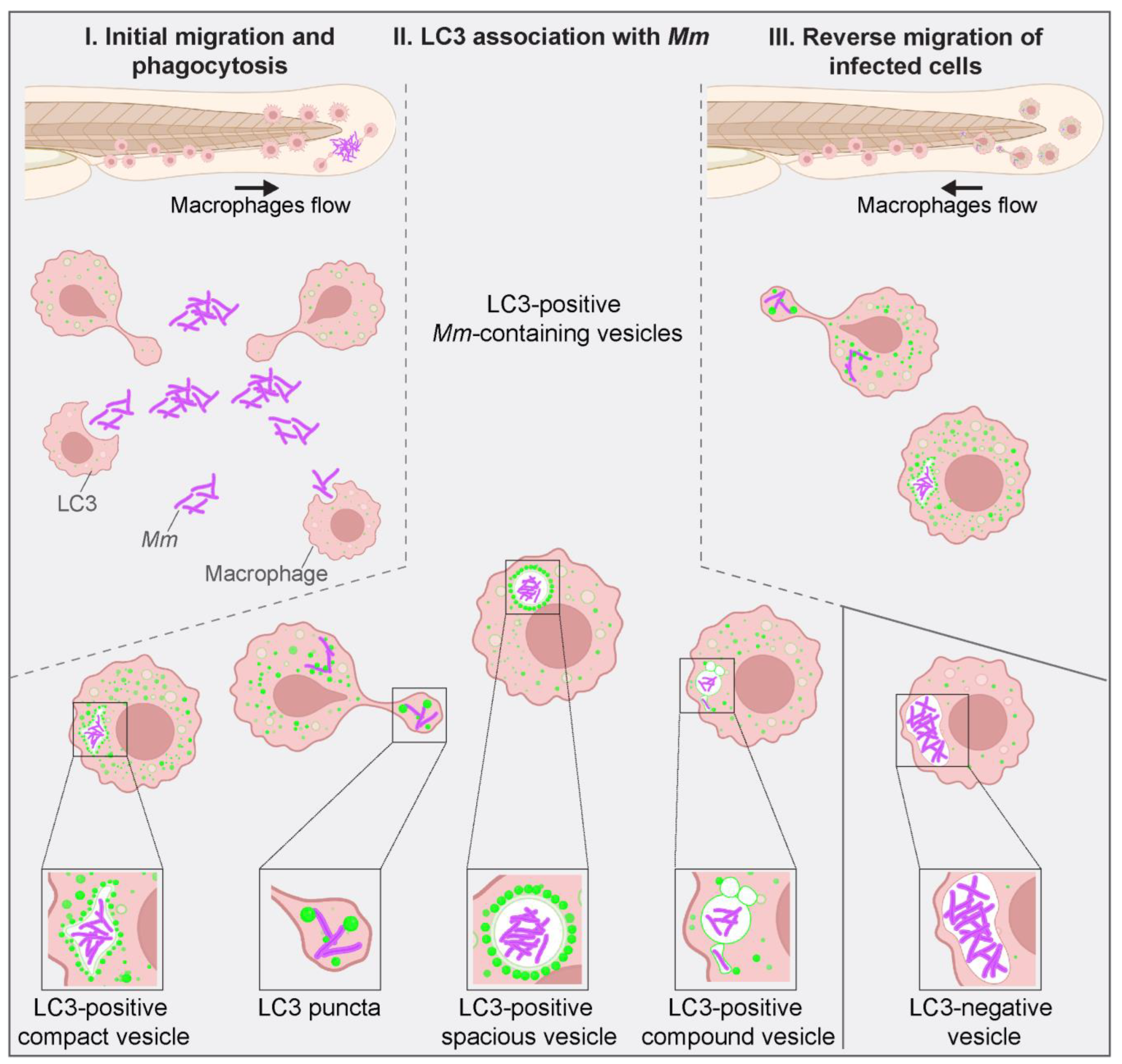
3.4. LC3-Positive Mm-Containing Vesicles Present Heterogeneous Dynamic Morphologies
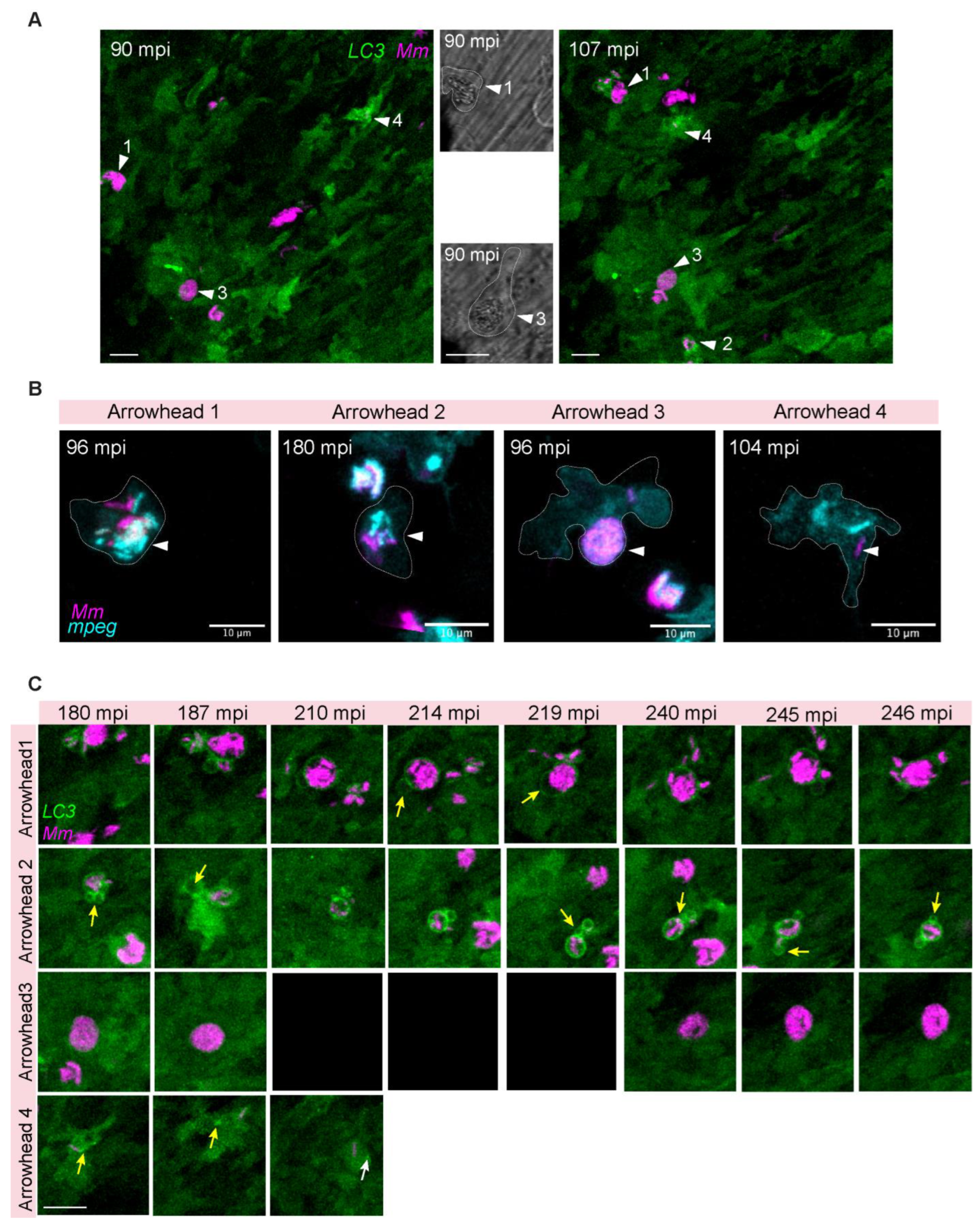
3.5. LC3-Mm Dynamics Occur Independent of the Bacterial Dose and Is Detected in Phagocytes Disseminating Mm Infection
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2022; WHO: Geneva, Switzerland, 2022. Available online: http://apps.who.int/bookorders (accessed on 30 January 2023).
- Bussi, C.; Gutierrez, M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019, 43, 341–361. [Google Scholar] [CrossRef]
- Kilinç, G.; Saris, A.; Ottenhoff, T.H.M.; Haks, M.C. Host-directed therapy to combat mycobacterial infections*. Immunol. Rev. 2021, 301, 62–83. [Google Scholar] [CrossRef]
- Russell, D.G.; Mwandumba, H.C.; Rhoades, E.E. Mycobacterium and the coat of many lipids. J. Cell Biol. 2002, 158, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Gröschel, M.I.; Sayes, F.; Simeone, R.; Majlessi, L.; Brosch, R. ESX secretion systems: Mycobacterial evolution to counter host immunity. Nat. Rev. Genet. 2016, 14, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Simeone, R.; Sayes, F.; Lawarée, E.; Brosch, R. Breaching the phagosome, the case of the tuberculosis agent. Cell. Microbiol. 2021, 23, e13344. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef]
- Deretic, V.; Delgado, M.; Vergne, I.; Master, S.; De Haro, S.; Ponpuak, M.; Singh, S. Autophagy in Immunity against Mycobacterium tuberculosis: A Model System to Dissect Immunological Roles of Autophagy. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 335, pp. 169–188. [Google Scholar] [CrossRef]
- Simeone, R.; Majlessi, L.; Enninga, J.; Brosch, R. Perspetives on mycobacterial vacuole-to-cytosol translocation: The importance of cytosolic access. Cell. Microbiol. 2016, 18, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Simeone, R.; Sayes, F.; Song, O.; Gröschel, M.I.; Brodin, P.; Brosch, R.; Majlessi, L. Cytosolic Access of Mycobacterium tuberculosis: Critical Impact of Phagosomal Acidification Control and Demonstration of Occurrence In Vivo. PLOS Pathog. 2015, 11, e1004650. [Google Scholar] [CrossRef]
- Watson, R.O.; Manzanillo, P.S.; Cox, J.S. Extracellular M. tuberculosis DNA Targets Bacteria for Autophagy by Activating the Host DNA-Sensing Pathway. Cell 2012, 150, 803–815. [Google Scholar] [CrossRef]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2016, 17, 811–819. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nature 2015, 17, 893–906. [Google Scholar] [CrossRef]
- Hooper, K.M.; Jacquin, E.; Li, T.; Goodwin, J.M.; Brumell, J.H.; Durgan, J.; Florey, O. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J. Cell Biol. 2022, 221, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.; Philips, J.A. LC3-associated phagocytosis: Host defense and microbial response. Curr. Opin. Immunol. 2019, 60, 81–90. [Google Scholar] [CrossRef]
- Boyle, K.B.; Randow, F. Rubicon swaps autophagy for LAP. Nature 2015, 17, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef]
- Köster, S.; Upadhyay, S.; Chandra, P.; Papavinasasundaram, K.; Yang, G.; Hassan, A.; Grigsby, S.J.; Mittal, E.; Park, H.S.; Jones, V.; et al. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. USA 2017, 114, E8711–E8720. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.M.; Ramakrishnan, L. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell. Microbiol. 2008, 10, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- van der Vaart, M.; Korbee, C.J.; Lamers, G.E.; Tengeler, A.C.; Hosseini, R.; Haks, M.C.; Ottenhoff, T.H.; Spaink, H.P.; Meijer, A.H. The DNA Damage-Regulated Autophagy Modulator DRAM1 Links Mycobacterial Recognition via TLR-MYD88 to Autophagic Defense. Cell Host Microbe 2014, 15, 753–767. [Google Scholar] [CrossRef]
- Cardenal-Muñoz, E.; Arafah, S.; López-Jiménez, A.T.; Kicka, S.; Falaise, A.; Bach, F.; Schaad, O.; King, J.S.; Hagedorn, M.; Soldati, T. Mycobacterium marinum antagonistically induces an autophagic response while repressing the autophagic flux in a TORC1- and ESX-1-dependent manner. PLOS Pathog. 2017, 13, e1006344. [Google Scholar] [CrossRef]
- van der Niet, S.; van Zon, M.; de Punder, K.; Grootemaat, A.; Rutten, S.; Moorlag, S.J.C.F.M.; Houben, D.; van der Sar, A.M.; Bitter, W.; Brosch, R.; et al. IL-1R1-Dependent Signals Improve Control of Cytosolic Virulent Mycobacteria In Vivo. Msphere 2021, 6, e00153-21. [Google Scholar] [CrossRef]
- Zhang, R.; Varela, M.; Vallentgoed, W.; Forn-Cuní, G.; Van Der Vaart, M.; Meijer, A.H. The selective autophagy receptors Optineurin and p62 are both required for zebrafish host resistance to mycobacterial infection. PLOS Pathog. 2019, 15, e1007329. [Google Scholar] [CrossRef]
- Zhang, R.; Varela, M.; Forn-Cuní, G.; Torraca, V.; van der Vaart, M.; Meijer, A.H. Deficiency in the autophagy modulator Dram1 exacerbates pyroptotic cell death of Mycobacteria-infected macrophages. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Robertson, T.F.; Huttenlocher, A. Real-time imaging of inflammation and its resolution: It’s apparent because it’s transparent*. Immunol. Rev. 2022, 306, 258–270. [Google Scholar] [CrossRef]
- He, C.; Bartholomew, C.R.; Zhou, W.; Klionsky, D.J. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy 2009, 5, 520–526. [Google Scholar] [CrossRef]
- Hosseini, R.; Lamers, G.E.; Hodzic, Z.; Meijer, A.H.; Schaaf, M.J.; Spaink, H.P. Correlative light and electron microscopy imaging of autophagy in a zebrafish infection model. Autophagy 2014, 10, 1844–1857. [Google Scholar] [CrossRef]
- Bernut, A.; Herrmann, J.-L.; Kissa, K.; Dubremetz, J.-F.; Gaillard, J.-L.; Lutfalla, G.; Kremer, L. Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation. Proc. Natl. Acad. Sci. USA 2014, 111, E943–E952. [Google Scholar] [CrossRef] [PubMed]
- Volkman, H.E.; Clay, H.; Beery, D.; Chang, J.; Sherman, D.R.; Ramakrishnan, L. Tuberculous granuloma Formation Is Enhanced by a Mycobacterium Virulence Determinant. PLoS Biol. 2004, 2, e367. [Google Scholar] [CrossRef] [PubMed]
- Takaki, K.; Davis, J.M.; Winglee, K.; Ramakrishnan, L. Evaluation of the pathogenesis and treatment of Mycobacterium marinum infection in zebrafish. Nat. Protoc. 2013, 8, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Benard, E.L.; van der Sar, A.; Ellett, F.; Lieschke, G.; Spaink, H.; Meijer, A.H. Infection of Zebrafish Embryos with Intracellular Bacterial Pathogens. J. Vis. Exp. 2012, 61, e3781. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Nguyen-Chi, M.; Laplace-Builhé, B.; Travnickova, J.; Luz-Crawford, P.; Tejedor, G.; Phan, Q.T.; Duroux-Richard, I.; Levraud, J.-P.; Kissa, K.; Lutfalla, G.; et al. Identification of polarized macrophage subsets in zebrafish. Elife 2015, 4, e07288. [Google Scholar] [CrossRef]
- Ku, J.W.K.; Chen, Y.; Lim, B.J.W.; Gasser, S.; Crasta, K.C.; Gan, Y.-H. Bacterial-induced cell fusion is a danger signal triggering cGAS–STING pathway via micronuclei formation. Proc. Natl. Acad. Sci. USA 2020, 117, 15923–15934. [Google Scholar] [CrossRef]
- Hosseini, R.; Lamers, G.E.M.; Soltani, H.M.; Meijer, A.H.; Spaink, H.P.; Schaaf, M.J.M. Efferocytosis and extrusion of leukocytes determine the progression of early mycobacterial pathogenesis. J. Cell Sci. 2016, 129, 3385–3395. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Bradfute, S.B.; Castillo, E.F.; Arko-Mensah, J.; Chauhan, S.; Jiang, S.; Mandell, M.; Deretic, V. Autophagy as an immune effector against tuberculosis. Curr. Opin. Microbiol. 2013, 16, 355–365. [Google Scholar] [CrossRef]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM Induces Autophagy to Eliminate Intracellular Mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.; Dekonenko, A.; Arko-Mensah, J.; Mandell, M.; Dupont, N.; Jiang, S.; Delgado-Vargas, M.; Timmins, G.; Bhattacharya, D.; Yang, H.; et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, E3168–E3176. [Google Scholar] [CrossRef] [PubMed]
- Strong, E.J.; Lee, S. Targeting Autophagy as a Strategy for Developing New Vaccines and Host-Directed Therapeutics Against Mycobacteria. Front. Microbiol. 2021, 11, 614313. [Google Scholar] [CrossRef]
- Kim, Y.S.; Silwal, P.; Kim, S.Y.; Yoshimori, T.; Jo, E.-K. Autophagy-activating strategies to promote innate defense against mycobacteria. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carinci, M.; Palumbo, L.; Pellielo, G.; Agyapong, E.D.; Morciano, G.; Patergnani, S.; Giorgi, C.; Pinton, P.; Rimessi, A. The Multifaceted Roles of Autophagy in Infectious, Obstructive, and Malignant Airway Diseases. Biomedicines 2022, 10, 1944. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S. Autophagy and bacterial clearance: A not so clear picture. Cell. Microbiol. 2012, 15, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S.; Cossart, P. Bacterial autophagy: Restriction or promotion of bacterial replication? Trends Cell Biol. 2012, 22, 283–291. [Google Scholar] [CrossRef]
- Cohen, S.B.; Gern, B.H.; Delahaye, J.L.; Adams, K.N.; Plumlee, C.R.; Winkler, J.K.; Sherman, D.R.; Gerner, M.Y.; Urdahl, K.B. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 2018, 24, 439–446.e4. [Google Scholar] [CrossRef]
- Cambier, C.J.; Takaki, K.K.; Larson, R.P.; Hernandez, R.E.; Tobin, D.M.; Urdahl, K.B.; Cosma, C.L.; Ramakrishnan, L. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 2014, 505, 218–222. [Google Scholar] [CrossRef]
- Corleis, B.; Dorhoi, A. Early dynamics of innate immunity during pulmonary tuberculosis. Immunol. Lett. 2020, 221, 56–60. [Google Scholar] [CrossRef]
- Bernard, E.M.; Fearns, A.; Bussi, C.; Santucci, P.; Peddie, C.J.; Lai, R.J.; Collinson, L.M.; Gutierrez, M.G. M. tuberculosis infection of human iPSDM reveals complex membrane dynamics during xenophagy evasion. J. Cell Sci. 2021, 134, jcs252973. [Google Scholar] [CrossRef] [PubMed]
- Heckmann, B.; Green, D.R. LC3-associated phagocytosis at a glance. J. Cell Sci. 2019, 132, jcs222984. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Sánchez, S.; Varela, M.; van der Vaart, M.; Meijer, A.H. Using Zebrafish to Dissect the Interaction of Mycobacteria with the Autophagic Machinery in Macrophages. Biology 2023, 12, 817. https://doi.org/10.3390/biology12060817
Muñoz-Sánchez S, Varela M, van der Vaart M, Meijer AH. Using Zebrafish to Dissect the Interaction of Mycobacteria with the Autophagic Machinery in Macrophages. Biology. 2023; 12(6):817. https://doi.org/10.3390/biology12060817
Chicago/Turabian StyleMuñoz-Sánchez, Salomé, Mónica Varela, Michiel van der Vaart, and Annemarie H. Meijer. 2023. "Using Zebrafish to Dissect the Interaction of Mycobacteria with the Autophagic Machinery in Macrophages" Biology 12, no. 6: 817. https://doi.org/10.3390/biology12060817
APA StyleMuñoz-Sánchez, S., Varela, M., van der Vaart, M., & Meijer, A. H. (2023). Using Zebrafish to Dissect the Interaction of Mycobacteria with the Autophagic Machinery in Macrophages. Biology, 12(6), 817. https://doi.org/10.3390/biology12060817