
Proline Isomerization: From the Chemistry and Biology to Therapeutic Opportunities

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Discovery and Study of Proline Isomerization in History: From Initial Observations to Modern Insights
3. Proline Isomerization Catalyzers: Peptidyl-Prolyl Isomerases
3.1. Cyclophilins
3.2. FK506 Binding Proteins (FKBPs)
3.3. Parvulins
4. Tying It Altogether: Showcase of Proline Isomerization as a Regulatory Mechanism in Cellular Response and Function
4.1. ATR Is Regulated by Proline Isomerization to Switch between Its Dual Role in Modulating Cell Death and DNA Damage Checkpoint Signaling
4.2. p53 Is Regulated by Proline Isomerization at Multiple Sites in Response to DNA Repair and Cellular Stress, Respectively
4.3. Itk Is Activated by Proline Isomerization in T-Cells
4.4. The Gate Function of 5-HT3 Receptor Is Regulated by Proline Isomerization
5. Role of Proline Isomerization in Human Disease
5.1. Autoimmune Disease
5.2. Cancer
5.3. Infectious Disease
5.4. Neurodegenerative Disease
6. Prolyl Isomerase Inhibitors
6.1. Cyclophilin Inhibitors
6.2. FKBP Inhibitors
6.3. Pin1 Inhibitors
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Willstatter, R. Synthesis of hygric acid. Berichte Dtsch. Chem. Ges. 1900, 33, 1160. [Google Scholar] [CrossRef]
- Fischer, E. Hydrolysis of casein by means of hydrochloric acid. J. Chem. Soc. 1901, 33, 151–176. [Google Scholar]
- Brandts, J.F.; Halvorson, H.R.; Brennan, M. Consideration of the Possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 1975, 14, 4953–4963. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.X.; Baldwin, R.L. Acid catalysis of the formation of the slow-folding species of RNase A: Evidence that the reaction is proline isomerization. Proc. Natl. Acad. Sci. USA 1978, 75, 4764–4768. [Google Scholar] [CrossRef]
- Lin, L.N.; Brandts, J.F. Role of cis-trans isomerism of the peptide bond in protease specificity. Kinetic studies on small proline-containing peptides and on polyproline. Biochemistry 1979, 18, 5037–5042. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.N.; Brandts, J.F. Kinetic mechanism for conformational transitions between poly-L-prolines I and II: A study utilizing the cis-trans specificity of a proline-specific protease. Biochemistry 1980, 19, 3055–3059. [Google Scholar] [CrossRef] [PubMed]
- Kendrew, J.C.; Bodo, G.; Dintzis, H.M.; Parrish, R.G.; Wyckoff, H.; Phillips, D.C. A three-dimensional model of the myoglobin molecule obtained by X-ray analysis. Nature 1958, 181, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.; Koenig, D.F.; Mair, G.A.; North, A.C.; Phillips, D.C.; Sarma, V.R. Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Angstrom resolution. Nature 1965, 206, 757–761. [Google Scholar] [CrossRef]
- Smith, J.L.; Hendrickson, W.A.; Honzatko, R.B.; Sheriff, S. Structural Heterogeneity in Protein Crystals. Biochemistry 1986, 25, 5018–5027. [Google Scholar] [CrossRef]
- Stewart, D.E.; Sarkar, A.; Wampler, J.E. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 1990, 214, 253–260. [Google Scholar] [CrossRef]
- Pal, D.; Chakrabarti, P. Cis peptide bonds in proteins: Residues involved, their conformations, interactions and locations. J. Mol. Biol. 1999, 294, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Reimer, U.; Fischer, G. Local structural changes caused by peptidyl-prolyl cis/trans isomerization in the native state of proteins. Biophys. Chem. 2002, 96, 203–212. [Google Scholar] [CrossRef]
- Pahlke, D.; Leitner, D.; Wiedemann, U.; Labudde, D. COPS—Cis/trans peptide bond conformation prediction of amino acids on the basis of secondary structure information. Bioinformatics 2005, 21, 685–686. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, A.H. Native state proline isomerization: An intrinsic molecular switch. Biochemistry 2003, 42, 9515–9524. [Google Scholar] [CrossRef] [PubMed]
- Grathwohl, C.; Wuthrich, K. Nmr-Studies of the Rates of Proline Cis-Trans Isomerization in Oligopeptides. Biopolymers 1981, 20, 2623–2633. [Google Scholar] [CrossRef]
- Sarkar, S.K.; Young, P.E.; Sullivan, C.E.; Torchia, D.A. Detection of cis and trans X-Pro peptide bonds in proteins by 13C NMR: Application to collagen. Proc. Natl. Acad. Sci. USA 1984, 81, 4800–4803. [Google Scholar] [CrossRef] [PubMed]
- Hodel, A.; Rice, L.M.; Simonson, T.; Fox, R.O.; Brunger, A.T. Proline Cis-Trans Isomerization in Staphylococcal Nuclease—Multi-Substate Free-Energy Perturbation Calculations. Protein Sci. 1995, 4, 636–654. [Google Scholar] [CrossRef] [PubMed]
- Higgins, K.A.; Craik, D.J.; Hall, J.G.; Andrews, P.R. Cis-trans isomerization of the proline residue in insulin studied by 13C NMR spectroscopy. Drug Des. Deliv. 1988, 3, 159–170. [Google Scholar] [PubMed]
- Chazin, W.J.; Kordel, J.; Drakenberg, T.; Thulin, E.; Brodin, P.; Grundstrom, T.; Forsen, S. Proline isomerism leads to multiple folded conformations of calbindin D9k: Direct evidence from two-dimensional 1H NMR spectroscopy. Proc. Natl. Acad. Sci. USA 1989, 86, 2195–2198. [Google Scholar] [CrossRef] [PubMed]
- Kordel, J.; Skelton, N.J.; Akke, M.; Chazin, W.J. High-resolution structure of calcium-loaded calbindin D9k. J. Mol. Biol. 1993, 231, 711–734. [Google Scholar] [CrossRef]
- Sebak, F.; Ecsedi, P.; Bermel, W.; Luy, B.; Nyitray, L.; Bodor, A. Selective (1) H(alpha) NMR Methods Reveal Functionally Relevant Proline cis/trans Isomers in Intrinsically Disordered Proteins: Characterization of Minor Forms, Effects of Phosphorylation, and Occurrence in Proteome. Angew. Chem. Int. Ed. Engl. 2022, 61, e202108361. [Google Scholar] [CrossRef] [PubMed]
- Hynes, T.R.; Fox, R.O. The Crystal-Structure of Staphylococcal Nuclease Refined at 1.7 a Resolution. Proteins-Struct. Funct. Genet. 1991, 10, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.A.; Thulin, E.; Forsen, S. Proline Cis-Trans Isomers in Calbindin D9k Observed by X-ray Crystallography. J. Mol. Biol. 1992, 223, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.A.; Dobson, C.M.; Kautz, R.A.; Hatfull, G.; Fox, R.O. Proline isomerism in staphylococcal nuclease characterized by NMR and site-directed mutagenesis. Nature 1987, 329, 266–268. [Google Scholar] [CrossRef]
- Shinoda, K.; Fujitani, H. Initiation of prolyl cis-trans isomerisation in the CDR-H3 loop of an antibody in response to antigen binding. Sci. Rep. 2017, 7, 16964. [Google Scholar] [CrossRef]
- Lietz, C.B.; Chen, Z.; Yun Son, C.; Pang, X.; Cui, Q.; Li, L. Multiple gas-phase conformations of proline-containing peptides: Is it always cis/trans isomerization? Analyst 2016, 141, 4863–4869. [Google Scholar] [CrossRef]
- Silzel, J.W.; Murphree, T.A.; Paranji, R.K.; Guttman, M.M.; Julian, R.R. Probing the Stability of Proline Cis/Trans Isomers in the Gas Phase with Ultraviolet Photodissociation. J. Am. Soc. Mass Spectrom. 2020, 31, 1974–1980. [Google Scholar] [CrossRef]
- Guttman, M.; Padte, N.N.; Huang, Y.; Yu, J.; Rocklin, G.J.; Weitzner, B.D.; Scian, M.; Ho, D.D.; Lee, K.K. The influence of proline isomerization on potency and stability of anti-HIV antibody 108. Sci. Rep. 2020, 10, 14313. [Google Scholar] [CrossRef]
- Wang, M.L.; Li, W.J.; Wang, M.L.; Xu, W.B. Support vector machines for prediction of peptidyl prolyl cis/trans isomerization. J. Pept. Res. 2004, 63, 23–28. [Google Scholar] [CrossRef]
- Song, J.; Burrage, K.; Yuan, Z.; Huber, T. Prediction of cis/trans isomerization in proteins using PSI-BLAST profiles and secondary structure information. BMC Bioinform. 2006, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Exarchos, K.P.; Exarchos, T.P.; Papaloukas, C.; Troganis, A.N.; Fotiadis, D.I. PBOND: Web server for the prediction of proline and non-proline cis/trans isomerization. Genom. Proteom. Bioinform. 2009, 7, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Al-Jarrah, O.Y.; Yoo, P.D.; Taha, K.; Muhaidat, S.; Shami, A.; Zaki, N. Randomized Subspace Learning for Proline Cis-Trans Isomerization Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Hanson, J.; Heffernan, R.; Paliwal, K.; Yang, Y.; Zhou, Y. Detecting Proline and Non-Proline Cis Isomers in Protein Structures from Sequences Using Deep Residual Ensemble Learning. J. Chem. Inf. Model. 2018, 58, 2033–2042. [Google Scholar] [CrossRef] [PubMed]
- Hanes, S.D. Prolyl isomerases in gene transcription. Biochim. Biophys. Acta-Gen. Subj. 2015, 1850, 2017–2034. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J. Cyclophilin—A Specific Cytosolic Binding-Protein for Cyclosporin-A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef]
- Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F.X. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989, 337, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Mikol, V.; Kallen, J.; Pflugl, G.; Walkinshaw, M.D. X-ray structure of a monomeric cyclophilin A-cyclosporin A crystal complex at 2.1 A resolution. J. Mol. Biol. 1993, 234, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Heitman, J. The cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Function-dependent clustering of orthologues and paralogues of cyclophilins. Proteins 2004, 56, 808–820. [Google Scholar] [CrossRef]
- Koletsky, A.J.; Harding, M.W.; Handschumacher, R.E. Cyclophilin: Distribution and variant properties in normal and neoplastic tissues. J. Immunol. 1986, 137, 1054–1059. [Google Scholar] [CrossRef]
- Schiene-Fischer, C.; Fischer, G.; Braun, M. Non-Immunosuppressive Cyclophilin Inhibitors. Angew. Chem.-Int. Ed. 2022, 61, e202201597. [Google Scholar] [CrossRef] [PubMed]
- Bergsma, D.J.; Eder, C.; Gross, M.; Kersten, H.; Sylvester, D.; Appelbaum, E.; Cusimano, D.; Livi, G.P.; McLaughlin, M.M.; Kasyan, K.; et al. The cyclophilin multigene family of peptidyl-prolyl isomerases. Characterization of three separate human isoforms. J. Biol. Chem. 1991, 266, 23204–23214. [Google Scholar] [CrossRef]
- Kieffer, L.J.; Seng, T.W.; Li, W.; Osterman, D.G.; Handschumacher, R.E.; Bayney, R.M. Cyclophilin-40, a protein with homology to the P59 component of the steroid receptor complex. Cloning of the cDNA and further characterization. J. Biol. Chem. 1993, 268, 12303–12310. [Google Scholar] [CrossRef] [PubMed]
- Schonbrunner, E.R.; Mayer, S.; Tropschug, M.; Fischer, G.; Takahashi, N.; Schmid, F.X. Catalysis of protein folding by cyclophilins from different species. J. Biol. Chem. 1991, 266, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Jankowski, W.; Sriram, G.; Rossi, P.; Shah, S.; Lee, K.B.; Cruz, L.A.; Rodriguez, A.J.; Birge, R.B.; Kalodimos, C.G. Cyclophilin A promotes cell migration via the Abl-Crk signaling pathway. Nat. Chem. Biol. 2016, 12, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Sowden, M.P.; Berk, B.C. Extracellular and Intracellular Cyclophilin A, Native and Post-Translationally Modified, Show Diverse and Specific Pathological Roles in Diseases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.H.; Yang, W.X.; Bai, X.Y.; Li, H.Q.; Li, H.Z.; Fan, W.H.; Zhang, H.; Liu, W.J.; Sun, L. Cyclophilin A is a key positive and negative feedback regulator within interleukin-6 trans-signaling pathway. Faseb J. 2021, 35, e21958. [Google Scholar] [CrossRef] [PubMed]
- Colgan, J.; Asmal, M.; Neagu, M.; Yu, B.; Schneidkraut, J.; Lee, Y.; Sokolskaja, E.; Andreotti, A.; Luban, J. Cyclophilin A regulates TCR signal strength in CD4+ T cells via a proline-directed conformational switch in Itk. Immunity 2004, 21, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Gegunde, S.; Alfonso, A.; Alvarino, R.; Alonso, E.; Gonzalez-Juanatey, C.; Botana, L.M. Crosstalk between cyclophilins and T lymphocytes in coronary artery disease. Exp. Cell Res. 2021, 400, 112514. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Hui, O.Y.; Lee, W.H.; et al. Structural and Biochemical Characterization of the Human Cyclophilin Family of Peptidyl-Prolyl Isomerases. PLoS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Harrison, S.C. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc. Natl. Acad. Sci. USA 2002, 99, 13522–13526. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.P.; Yamashita, M.M.; Sintchak, M.D.; Rotstein, S.H.; Murcko, M.A.; Boger, J.; Thomson, J.A.; Fitzgibbon, M.J.; Black, J.R.; Navia, M.A. Comparative X-Ray Structures of the Major Binding-Protein for the Immunosuppressant Fk506 (Tacrolimus) in Unliganded Form and in Complex with Fk506 and Rapamycin. Acta Crystallogr. Sect. D-Biol. Crystallogr. 1995, 51, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Daum, S.; Wildemann, D.; Zhou, X.Z.; Verdecia, M.A.; Bowman, M.E.; Lucke, C.; Hunter, T.; Lu, K.P.; Fischer, G.; et al. Structural basis for high-affinity peptide inhibition of human Pin1. ACS Chem. Biol. 2007, 2, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Siekierka, J.J.; Hung, S.H.; Poe, M.; Lin, C.S.; Sigal, N.H. A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 1989, 341, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.W.; Galat, A.; Uehling, D.E.; Schreiber, S.L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 1989, 341, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Choi, J.; Clardy, J. Refined structure of the FKBP12-rapamycin-FRB ternary complex at 2.2 angstrom resolution. Acta Crystallogr. Sect. D-Struct. Biol. 1999, 55, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.P.; Kim, J.L.; Kim, E.E.; Sintchak, M.D.; Thomson, J.A.; Fitzgibbon, M.J.; Fleming, M.A.; Caron, P.R.; Hsiao, K.; Navia, M.A. X-ray Structure of Calcineurin Inhibited by the Immunophilin Immunosuppressant Fkbp12-Fk506 Complex. Cell 1995, 82, 507–522. [Google Scholar] [CrossRef]
- Kolos, J.M.; Voll, A.M.; Bauder, M.; Hausch, F. FKBP Ligands—Where We Are and Where to Go? Front. Pharmacol. 2018, 9, 1425. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Aumuller, T. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. Rev. Physiol. Biochem. Pharmacol. 2003, 148, 105–150. [Google Scholar] [PubMed]
- Kang, C.B.; Hong, Y.; Dhe-Paganon, S.; Yoon, H.S. FKBP family proteins: Immunophilins with versatile biological functions. Neurosignals 2008, 16, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ghartey-Kwansah, G.; Li, Z.; Feng, R.; Wang, L.; Zhou, X.; Chen, F.Z.; Xu, M.M.; Jones, O.; Mu, Y.; Chen, S.; et al. Comparative analysis of FKBP family protein: Evaluation, structure, and function in mammals and Drosophila melanogaster. BMC Dev. Biol. 2018, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Jiang, Y. FK506-Binding Proteins and Their Diverse Functions. Curr. Mol. Pharmacol. 2015, 9, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Rahfeld, J.U.; Rucknagel, K.P.; Schelbert, B.; Ludwig, B.; Hacker, J.; Mann, K.; Fischer, G. Confirmation of The Existence of A 3rd Family Among Peptidyl-Prolyl Cis/Trans Isomerases—Amino-Acid-Sequence And Recombinant Production of Parvulin. FEBS Lett. 1994, 352, 180–184. [Google Scholar] [CrossRef]
- Rahfeld, J.U.; Schierhorn, A.; Mann, K.; Fischer, G. A Novel Peptidyl-Prolyl Cis/Trans Isomerase from Escherichia coli. FEBS Lett. 1994, 343, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Hanes, S.D.; Hunter, T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 1996, 380, 544–547. [Google Scholar] [PubMed]
- Uchida, T.; Fujimori, F.; Tradler, T.; Fischer, G.; Rahfeld, J.U. Identification and characterization of a 14 kDa human protein as a novel parvulin-like peptidyl prolyl cis/trans isomerase. FEBS Lett. 1999, 446, 278–282. [Google Scholar] [CrossRef]
- Rulten, S.; Thorpe, J.; Kay, J. Identification of eukaryotic parvulin homologues: A new subfamily of peptidylprolyl cis-trans isomerases. Biochem. Biophys. Res. Commun. 1999, 259, 557–562. [Google Scholar] [CrossRef]
- Mueller, J.W.; Kessler, D.; Neumann, D.; Stratmann, T.; Papatheodorou, P.; Hartmann-Fatu, C.; Bayer, P. Characterization of novel elongated Parvulin isoforms that are ubiquitously expressed in human tissues and originate from alternative transcription initiation. BMC Mol. Biol. 2006, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Matena, A.; Rehic, E.; Honig, D.; Kamba, B.; Bayer, P. Structure and function of the human parvulins Pin1 and Par14/17. Biol. Chem. 2018, 399, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Lu, K.P.; Hunter, T.; Noel, J.P. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell 1997, 89, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B.; Schutkowski, M.; Shen, M.; Zhou, X.Z.; Stukenberg, P.T.; Rahfeld, J.U.; Xu, J.; Kuang, J.; Kirschner, M.W.; Fischer, G.; et al. Sequence-specific and phosphorylation-dependent proline isomerization: A potential mitotic regulatory mechanism. Science 1997, 278, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.W.; Bayer, P. Small family with key contacts: par14 and par17 parvulin proteins, relatives of pin1, now emerge in biomedical research. Perspect. Med. Chem. 2008, 2, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Takamiya, M.; Takahashi, M.; Miyashita, H.; Ikeda, H.; Terada, T.; Matsuo, Y.; Shirouzu, M.; Yokoyama, S.; Fujimori, F.; et al. Pin1 and Par14 peptidyl prolyl isomerase inhibitors block cell proliferation. Chem. Biol. 2003, 10, 15–24. [Google Scholar] [CrossRef]
- Burgardt, N.I.; Schmidt, A.; Manns, A.; Schutkowski, A.; Jahreis, G.; Lin, Y.J.; Schulze, B.; Masch, A.; Lucke, C.; Weiwad, M. Parvulin 17-catalyzed Tubulin Polymerization Is Regulated by Calmodulin in a Calcium-dependent Manner. J. Biol. Chem. 2015, 290, 16708–16722. [Google Scholar] [CrossRef]
- Kim, K. PPIases Par14/Par17 Affect HBV Replication in Multiple Ways. Viruses 2023, 15, 457. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef]
- El Boustani, M.; De Stefano, L.; Caligiuri, I.; Mouawad, N.; Granchi, C.; Canzonieri, V.; Tuccinardi, T.; Giordano, A.; Rizzolio, F. A Guide to PIN1 Function and Mutations across Cancers. Front. Pharmacol. 2019, 9, 1477. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Y.R.; Yang, H.Y.; Li, X.Z.; Jie, M.M.; Hu, C.J.; Wu, Y.Y.; Yang, S.M.; Yang, Y.B. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Baltimore, D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003, 17, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Hilton, B.A.; Li, Z.; Musich, P.R.; Wang, H.; Cartwright, B.M.; Serrano, M.; Zhou, X.Z.; Lu, K.P.; Zou, Y. ATR Plays a Direct Antiapoptotic Role at Mitochondria, which Is Regulated by Prolyl Isomerase Pin1. Mol. Cell 2015, 60, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Makinwa, Y.; Cartwright, B.M.; Musich, P.R.; Li, Z.K.; Biswas, H.; Zou, Y. PP2A Regulates Phosphorylation-Dependent Isomerization of Cytoplasmic and Mitochondrial-Associated ATR by Pin1 in DNA Damage Responses. Front. Cell Dev. Biol. 2020, 8, 813. [Google Scholar] [CrossRef] [PubMed]
- Biswas, H.; Zhao, S.J.; Makinwa, Y.; Bassett, J.S.; Musich, P.R.; Liu, J.Y.; Zou, Y. Prolyl Isomerization-Mediated Conformational Changes Define ATR Subcellular Compartment-Specific Functions. Front. Cell Dev. Biol. 2022, 10, 826576. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.H.; Liu, M.J.; Tian, Y.; Wu, Z.H.; Hao, Y.H.; Song, L.; Qin, Z.Y.; Ding, C.; Wang, H.W.; Wang, J.W.; et al. Cryo-EM structure of human ATR-ATRIP complex. Cell Res. 2018, 28, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Makinwa, Y.; Musich, P.R.; Zou, Y. Phosphorylation-Dependent Pin1 Isomerization of ATR: Its Role in Regulating ATR’s Anti-apoptotic Function at Mitochondria, and the Implications in Cancer. Front. Cell Dev. Biol. 2020, 8, 281. [Google Scholar] [CrossRef]
- Liu, M.D.; Zeng, T.L.; Zhang, X.; Liu, C.Y.; Wu, Z.H.; Yao, L.M.; Xie, C.C.; Xia, H.; Lin, Q.; Xie, L.P.; et al. ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat. Commun. 2018, 9, 4139. [Google Scholar] [CrossRef]
- Mallis, R.J.; Brazin, K.N.; Fulton, D.B.; Andreotti, A.H. Structural characterization of a proline-driven conformational switch within the Itk SH2 domain. Nat. Struct. Biol. 2002, 9, 900–905. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef]
- Zheng, H.W.; You, H.; Zhou, X.Z.; Murray, S.A.; Uchida, T.; Wult, G.; Gu, L.; Tang, X.R.; Lu, K.P.; Xiao, Z.X.J. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002, 419, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.M.; Liou, Y.C.; Ryo, A.; Lee, S.W.; Lu, K.P. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J. Biol. Chem. 2002, 277, 47976–47979. [Google Scholar] [CrossRef] [PubMed]
- Zacchi, P.; Gostissa, M.; Uchida, T.; Salvagno, C.; Avolio, F.; Volinia, S.; Ronai, Z.; Blandino, G.; Schneider, C.; Del Sal, G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002, 419, 853–857. [Google Scholar] [CrossRef]
- Zhan, Y.A.; Ytreberg, F.M. The cis conformation of proline leads to weaker binding of a p53 peptide to MDM2 compared to trans. Arch. Biochem. Biophys. 2015, 575, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Follis, A.V.; Llambi, F.; Merritt, P.; Chipuk, J.E.; Green, D.R.; Kriwacki, R.W. Pin1-Induced Proline Isomerization in Cytosolic p53 Mediates BAX Activation and Apoptosis. Mol. Cell 2015, 59, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Kumutima, J.; Yao, X.Q.; Hamelberg, D. p53 Is Potentially Regulated by Cyclophilin D in the Triple-Proline Loop of the DNA Binding Domain. Biochemistry 2021, 60, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, X.Y.; Blayney, A.; Zhang, Y.M.; Gandy, L.; Mirsky, P.O.; Smith, N.; Zhang, F.M.; Linhardt, R.J.; Chen, J.H.; et al. Intrinsically Disordered N-terminal Domain (NTD) of p53 Interacts with Mitochondrial PTP Regulator Cyclophilin D. J. Mol. Biol. 2022, 434, 167552. [Google Scholar] [CrossRef]
- Karch, J.; Molkentin, J.D. Is p53 the Long-Sought Molecular Trigger for Cyclophilin D-Regulated Mitochondrial Permeability Transition Pore Formation and Necrosis? Circ. Res. 2012, 111, 1258–1260. [Google Scholar] [CrossRef] [PubMed]
- Baum, N.; Schiene-Fischer, C.; Frost, M.; Schumann, M.; Sabapathy, K.; Ohlenschlager, O.; Grosse, F.; Schlott, B. The prolyl cis/trans isomerase cyclophilin 18 interacts with the tumor suppressor p53 and modifies its functions in cell cycle regulation and apoptosis. Oncogene 2009, 28, 3915–3925. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, A.H.; Schwartzberg, P.L.; Joseph, R.E.; Berg, L.J. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect. Biol. 2010, 2, a002287. [Google Scholar] [CrossRef]
- Brazin, K.N.; Mallis, R.J.; Fulton, D.B.; Andreotti, A.H. Regulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclophilin, A. Proc. Natl. Acad. Sci. USA 2002, 99, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Bunnell, S.C.; Diehn, M.; Yaffe, M.B.; Findell, P.R.; Cantley, L.C.; Berg, L.J. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J. Biol. Chem. 2000, 275, 2219–2230. [Google Scholar] [CrossRef]
- Pletneva, E.V.; Sundd, M.; Fulton, D.B.; Andreotti, A.H. Molecular details of Itk activation by prolyl isomerization and phospholigand binding: The NMR structure of the Itk SH2 domain bound to a phosphopeptide. J. Mol. Biol. 2006, 357, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Severin, A.; Joseph, R.E.; Boyken, S.; Fulton, D.B.; Andreotti, A.H. Proline Isomerization Preorganizes the Itk SH2 Domain for Binding to the Itk SH3 Domain. J. Mol. Biol. 2009, 387, 726–743. [Google Scholar] [CrossRef] [PubMed]
- Breheny, P.J.; Laederach, A.; Fulton, D.B.; Andreotti, A.H. Ligand specificity modulated by prolyl imide bond cis/trans isomerization in the Itk SH2 domain: A quantitative NMR study. J. Am. Chem. Soc. 2003, 125, 15706–15707. [Google Scholar] [CrossRef] [PubMed]
- Lynagh, T.; Lynch, J.W. Ivermectin binding sites in human and invertebrate Cys-loop receptors. Trends Pharmacol. Sci. 2012, 33, 432–441. [Google Scholar] [CrossRef]
- Thompson, A.J.; Lummis, S.C. 5-HT3 receptors. Curr. Pharm. Des. 2006, 12, 3615–3630. [Google Scholar] [CrossRef] [PubMed]
- Deane, C.M.; Lummis, S.C.R. The role and predicted propensity of conserved proline residues in the 5-HT3 receptor. J. Biol. Chem. 2001, 276, 37962–37966. [Google Scholar] [CrossRef] [PubMed]
- Lummis, S.C.; Beene, D.L.; Lee, L.W.; Lester, H.A.; Broadhurst, R.W.; Dougherty, D.A. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature 2005, 438, 248–252. [Google Scholar] [CrossRef]
- Crnjar, A.; Comitani, F.; Hester, W.; Molteni, C. Trans-Cis Proline Switches in a Pentameric Ligand-Gated Ion Channel: How They Are Affected by and How They Affect the Biomolecular Environment. J. Phys. Chem. Lett. 2019, 10, 694–700. [Google Scholar] [CrossRef]
- Paulsen, I.M.; Martin, I.L.; Dunn, S.M.J. Isomerization of the proline in the M2-M3 linker is not required for activation of the human 5-HT(3)A receptor. J. Neurochem. 2009, 110, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Mosesso, R.; Dougherty, D.A.; Lummis, S.C.R. Proline Residues in the Transmembrane/Extracellular Domain Interface Loops Have Different Behaviors in 5-HT3 and nACh Receptors. ACS Chem. Neurosci. 2019, 10, 3327–3333. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mork, S.; Bo, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Bjartmar, C.; Trapp, B.D. Axonal and neuronal degeneration in multiple sclerosis: Mechanisms and functional consequences. Curr. Opin. Neurol. 2001, 14, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Metwally, E.; Al-Abbadi, H.A.; Hashem, M.A.; Mahmoud, Y.K.; Ahmed, E.A.; Maaty, A.I.; Helal, I.E.; Ahmed, M.F. Selective Calpain Inhibition Improves Functional and Histopathological Outcomes in a Canine Spinal Cord Injury Model. Int. J. Mol. Sci. 2022, 23, 11772. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, X.; Fowlkes, J.; Rahder, M.; Stem, K.; et al. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar] [CrossRef] [PubMed]
- Warne, J.; Pryce, G.; Hill, J.M.; Shi, X.; Lenneras, F.; Puentes, F.; Kip, M.; Hilditch, L.; Walker, P.; Simone, M.I.; et al. Selective Inhibition of the Mitochondrial Permeability Transition Pore Protects against Neurodegeneration in Experimental Multiple Sclerosis. J. Biol. Chem. 2016, 291, 4356–4373. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.Z.; Wu, Y.B.; Xue, Z.Y.; Zhang, K.; Zhang, R.X. The therapeutic effects of the peptidyl-prolyl cis/trans isomerase Pin1 inhibitor juglone on animal-model experimental autoimmune encephalomyelitis. J. Physiol. Pharmacol. 2021, 72, 195–202. [Google Scholar]
- Goverman, J.; Woods, A.; Larson, L.; Weiner, L.P.; Hood, L.; Zaller, D.M. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell 1993, 72, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Polverini, E.; Rangaraj, G.; Libich, D.S.; Boggs, J.M.; Harauz, G. Binding of the proline-rich segment of myelin basic protein to SH3 domains: Spectroscopic, microarray, and modeling studies of ligand conformation and effects of posttranslational modifications. Biochemistry 2008, 47, 267–282. [Google Scholar] [CrossRef] [PubMed]
- Vassall, K.A.; Jenkins, A.D.; Bamm, V.V.; Harauz, G. Thermodynamic analysis of the disorder-to-alpha-helical transition of 18.5-kDa myelin basic protein reveals an equilibrium intermediate representing the most compact conformation. J. Mol. Biol. 2015, 427, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Vakilian, M. A review on the effect of prolyl isomerization on immune response aberration and hypersensitivity reactions: A unifying hypothesis. Clin. Immunol. 2022, 234, 108896. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Burow, A.; Kurrer, M.; Lang, P.A.; Recher, M. The role of the innate immune response in autoimmune disease. J. Autoimmun. 2007, 29, 206–212. [Google Scholar] [CrossRef]
- Sim, T.M.; Ong, S.J.; Mak, A.; Tay, S.H. Type I Interferons in Systemic Lupus Erythematosus: A Journey from Bench to Bedside. Int. J. Mol. Sci. 2022, 23, 2505. [Google Scholar] [CrossRef]
- Preble, O.T.; Black, R.J.; Friedman, R.M.; Klippel, J.H.; Vilcek, J. Systemic lupus erythematosus: Presence in human serum of an unusual acid-labile leukocyte interferon. Science 1982, 216, 429–431. [Google Scholar] [CrossRef]
- Lipsky, P.E. Systemic lupus erythematosus: An autoimmune disease of B cell hyperactivity. Nat. Immunol. 2001, 2, 764–766. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef]
- Tun-Kyi, A.; Finn, G.; Greenwood, A.; Nowak, M.; Lee, T.H.; Asara, J.M.; Tsokos, G.C.; Fitzgerald, K.; Israel, E.; Li, X.; et al. Essential role for the prolyl isomerase Pin1 in Toll-like receptor signaling and type I interferon-mediated immunity. Nat. Immunol. 2011, 12, 733–741. [Google Scholar] [CrossRef]
- Wei, S.; Yoshida, N.; Finn, G.; Kozono, S.; Nechama, M.; Kyttaris, V.C.; Zhen Zhou, X.; Tsokos, G.C.; Ping Lu, K. Pin1-Targeted Therapy for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016, 68, 2503–2513. [Google Scholar] [CrossRef] [PubMed]
- Takeno, M.; Gunn, J. A novel role of peptidyl-prolyl isomerase-1 as inducer of IL-6 expression in systemic lupus erythematosus. Am. J. Biomed. 2015, 3, 439–450. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Tackey, E.; Lipsky, P.E.; Illei, G.G. Rationale for interleukin-6 blockade in systemic lupus erythematosus. Lupus 2004, 13, 339–343. [Google Scholar] [CrossRef]
- Lufei, C.; Koh, T.H.; Uchida, T.; Cao, X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene 2007, 26, 7656–7664. [Google Scholar] [CrossRef] [PubMed]
- Billich, A.; Winkler, G.; Aschauer, H.; Rot, A.; Peichl, P. Presence of cyclophilin A in synovial fluids of patients with rheumatoid arthritis. J. Exp. Med. 1997, 185, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, W.J.; Jeon, S.T.; Koh, E.M.; Cha, H.S.; Ahn, K.S.; Lee, W.H. Cyclophilin A may contribute to the inflammatory processes in rheumatoid arthritis through induction of matrix degrading enzymes and inflammatory cytokines from macrophages. Clin. Immunol. 2005, 116, 217–224. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, N.; Zhou, J.; Chen, Z.N.; Zhu, P. Cyclophilin A up-regulates MMP-9 expression and adhesion of monocytes/macrophages via CD147 signalling pathway in rheumatoid arthritis. Rheumatology 2008, 47, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, C.H.; Wang, Y.H.; Fan, C.M.; Zhu, P. The role of CyPA in chemotaxis of neutrophil in rheumatoid arthritis and secretion of interleukin-8. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2009, 25, 423–425. [Google Scholar]
- Wang, L.; Wang, C.H.; Jia, J.F.; Ma, X.K.; Li, Y.; Zhu, H.B.; Tang, H.; Chen, Z.N.; Zhu, P. Contribution of cyclophilin A to the regulation of inflammatory processes in rheumatoid arthritis. J. Clin. Immunol. 2010, 30, 24–33. [Google Scholar] [CrossRef]
- Wang, C.H.; Dai, J.Y.; Wang, L.; Jia, J.F.; Zheng, Z.H.; Ding, J.; Chen, Z.N.; Zhu, P. Expression of CD147 (EMMPRIN) on neutrophils in rheumatoid arthritis enhances chemotaxis, matrix metalloproteinase production and invasiveness of synoviocytes. J. Cell Mol. Med. 2011, 15, 850–860. [Google Scholar] [CrossRef]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef]
- Bao, L.; Kimzey, A.; Sauter, G.; Sowadski, J.M.; Lu, K.P.; Wang, D.G. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol. 2004, 164, 1727–1737. [Google Scholar] [CrossRef]
- Miyashita, H.; Uchida, T.; Mori, S.; Echigo, S.; Motegi, K. Expression status of Pin1 and cyclins in oral squamous cell carcinoma: Pin1 correlates with Cyclin D1 mRNA expression and clinical significance of cyclins. Oncol. Rep. 2003, 10, 1045–1048. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.W.; Tsai, C.H.; Hsiao, M.; Tseng, C.J.; Ger, L.P.; Lee, K.H.; Lu, P.J. Pin1 overexpression is associated with poor differentiation and survival in oral squamous cell carcinoma. Oncol. Rep. 2009, 21, 1097–1104. [Google Scholar] [PubMed]
- Zhang, Z.; Yu, W.; Zheng, M.; Liao, X.; Wang, J.; Yang, D.; Lu, W.; Wang, L.; Zhang, S.; Liu, H.; et al. Pin1 inhibition potently suppresses gastric cancer growth and blocks PI3K/AKT and Wnt/beta-catenin oncogenic pathways. Mol. Carcinog. 2019, 58, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Lin, Y.M.; Kozono, S.; Herbert, M.K.; Li, X.; Yuan, X.; Guo, J.; Guo, Y.; Tang, M.; Lin, J.; et al. Pin1 inhibition exerts potent activity against acute myeloid leukemia through blocking multiple cancer-driving pathways. J. Hematol. Oncol. 2018, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.J.; Cho, Y.G.; Park, Y.G.; Nam, S.W.; Kim, S.Y.; Lee, S.H.; Yoo, N.J.; Lee, J.Y.; Park, W.S. Pin1 overexpression in colorectal cancer and its correlation with aberrant beta-catenin expression. World J. Gastroenterol. 2005, 11, 5006–5009. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, J.; Arai, T.; Ikeda, S.; Kumagai, J.; Uetake, H.; Sugihara, K. High Pin1 expression is associated with tumor progression in colorectal cancer. J. Surg. Oncol. 2006, 94, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Park, B.H.; Park, J.H.; Jang, K.Y.; Park, H.S.; Wagle, S.; Lee, K.B.; Kim, J.R. Overexpression of the prolyl isomerase PIN1 promotes cell growth in osteosarcoma cells. Oncol. Rep. 2013, 29, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Koikawa, K.; Kibe, S.; Suizu, F.; Sekino, N.; Kim, N.; Manz, T.D.; Pinch, B.J.; Akshinthala, D.; Verma, A.; Gaglia, G.; et al. Targeting Pin1 renders pancreatic cancer eradicable by synergizing with immunochemotherapy. Cell 2021, 184, 4753–4771.e27. [Google Scholar] [CrossRef]
- Naito, M.; Ikeda, K.; Aoyama, S.; Kanamoto, M.; Akasaka, Y.; Kido, Y.; Nakanishi, M.; Kanna, M.; Yamamotoya, T.; Matsubara, A.; et al. Par14 interacts with the androgen receptor, augmenting both its transcriptional activity and prostate cancer proliferation. Cancer Med. 2023, 12, 8464–8475. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, T.; Cluning, C.; Ward, B.K. Steroid Receptor-Associated Immunophilins: A Gateway to Steroid Signalling. Clin. Biochem. Rev. 2015, 36, 31–52. [Google Scholar] [PubMed]
- Periyasamy, S.; Warrier, M.; Tillekeratne, M.P.; Shou, W.; Sanchez, E.R. The immunophilin ligands cyclosporin A and FK506 suppress prostate cancer cell growth by androgen receptor-dependent and -independent mechanisms. Endocrinology 2007, 148, 4716–4726. [Google Scholar] [CrossRef]
- Habara, M.; Sato, Y.; Goshima, T.; Sakurai, M.; Imai, H.; Shimizu, H.; Katayama, Y.; Hanaki, S.; Masaki, T.; Morimoto, M.; et al. FKBP52 and FKBP51 differentially regulate the stability of estrogen receptor in breast cancer. Proc. Natl. Acad. Sci. USA 2022, 119, e2110256119. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Qin, X.; Fang, J.; Tang, Y.; Fan, Y. Multi-Omics Analysis of the Expression and Prognosis for FKBP Gene Family in Renal Cancer. Front. Oncol. 2021, 11, 697534. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cui, X.; Feng, L.; Han, Z.; Peng, D.; Fu, W.; Xing, Y. The deficiency of FKBP-5 inhibited hepatocellular progression by increasing the infiltration of distinct immune cells and inhibiting obesity-associated gut microbial metabolite. J. Gastrointest. Oncol. 2021, 12, 711–721. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, S.; Zhang, M.; Liu, X.; Ju, G.; Hou, J. A Novel Biomarker, FKBP10, for Poor Prognosis Prediction in Patients with Clear Cell Renal Cell Carcinoma. Evid.-Based Complement. Altern. Med. 2022, 2022, 5490644. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.; Finn, G.; Suizu, F.; Lu, K.P. Phosphorylation-specific prolyl isomerization: Is there an underlying theme? Nat. Cell Biol. 2005, 7, 435–441. [Google Scholar] [CrossRef]
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478. [Google Scholar] [CrossRef]
- Zannini, A.; Rustighi, A.; Campaner, E.; Del Sal, G. Oncogenic Hijacking of the PIN1 Signaling Network. Front. Oncol. 2019, 9, 94. [Google Scholar] [CrossRef]
- Wulf, G.M.; Ryo, A.; Wulf, G.G.; Lee, S.W.; Niu, T.H.; Petkova, V.; Lu, K.P. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 2001, 20, 3459–3472. [Google Scholar] [CrossRef]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Im, C.Y.; Min, S.H. Function of PIN1 in Cancer Development and Its Inhibitors as Cancer Therapeutics. Front. Cell Dev. Biol. 2020, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Wulf, G.; Garg, P.; Liou, Y.C.; Iglehart, D.; Lu, K.P. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004, 23, 3397–3407. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Zhou, F.; Wan, J.; Hang, J.; Chen, Z.; Li, B.; Zhang, C.; Shao, K.; Jiang, P.; Shi, S.; et al. Pin1 expression contributes to lung cancer: Prognosis and carcinogenesis. Cancer Biol. Ther. 2010, 9, 111–119. [Google Scholar] [CrossRef]
- Franciosa, G.; Diluvio, G.; Gaudio, F.D.; Giuli, M.V.; Palermo, R.; Grazioli, P.; Campese, A.F.; Talora, C.; Bellavia, D.; D’Amati, G.; et al. Prolyl-isomerase Pin1 controls Notch3 protein expression and regulates T-ALL progression. Oncogene 2016, 35, 4741–4751. [Google Scholar] [CrossRef]
- D’Artista, L.; Bisso, A.; Piontini, A.; Doni, M.; Verrecchia, A.; Kress, T.R.; Morelli, M.J.; Del Sal, G.; Amati, B.; Campaner, S. Pin1 is required for sustained B cell proliferation upon oncogenic activation of Myc. Oncotarget 2016, 7, 21786–21798. [Google Scholar] [CrossRef]
- Karna, S.K.L.; Ahmad, F.; Lone, B.A.; Pokharel, Y.R. Knockdown of PTOV1 and PIN1 exhibit common phenotypic anti-cancer effects in MDA-MB-231 cells. PLoS ONE 2019, 14, e0211658. [Google Scholar] [CrossRef]
- Han, C.H.; Lu, J.; Wei, Q.; Bondy, M.L.; Brewster, A.M.; Yu, T.K.; Buchholz, T.A.; Arun, B.K.; Wang, L.E. The functional promoter polymorphism (−842G>C) in the PIN1 gene is associated with decreased risk of breast cancer in non-Hispanic white women 55 years and younger. Breast Cancer Res. Treat. 2010, 122, 243–249. [Google Scholar] [CrossRef]
- Li, Q.; Dong, Z.; Lin, Y.; Jia, X.; Li, Q.; Jiang, H.; Wang, L.; Gao, Y. The rs2233678 polymorphism in PIN1 promoter region reduced cancer risk: A meta-analysis. PLoS ONE 2013, 8, e68148. [Google Scholar] [CrossRef] [PubMed]
- Suizu, F.; Ryo, A.; Wulf, G.; Lim, J.; Lu, K.P. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol. Cell Biol. 2006, 26, 1463–1479. [Google Scholar] [CrossRef] [PubMed]
- Namgoong, G.M.; Khanal, P.; Cho, H.G.; Lim, S.C.; Oh, Y.K.; Kang, B.S.; Shim, J.H.; Yoo, J.C.; Choi, H.S. The prolyl isomerase Pin1 induces LC-3 expression and mediates tamoxifen resistance in breast cancer. J. Biol. Chem. 2010, 285, 23829–23841. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.R.; Choi, H.S.; Yang, J.W.; Park, B.C.; Kim, J.A.; Kang, K.W. Enhancement of vascular endothelial growth factor-mediated angiogenesis in tamoxifen-resistant breast cancer cells: Role of Pin1 overexpression. Mol. Cancer Ther. 2009, 8, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Liu, Z.; Shi, F.; Wang, J. Pin1 modulates chemo-resistance by up-regulating FoxM1 and the involvements of Wnt/beta-catenin signaling pathway in cervical cancer. Mol. Cell Biochem. 2016, 413, 179–187. [Google Scholar] [CrossRef]
- Nishi, M.; Akutsu, H.; Masui, S.; Kondo, A.; Nagashima, Y.; Kimura, H.; Perrem, K.; Shigeri, Y.; Toyoda, M.; Okayama, A.; et al. A distinct role for Pin1 in the induction and maintenance of pluripotency. J. Biol. Chem. 2011, 286, 11593–11603. [Google Scholar] [CrossRef]
- Sinars, C.R.; Cheung-Flynn, J.; Rimerman, R.A.; Scammell, J.G.; Smith, D.F.; Clardy, J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 868–873. [Google Scholar] [CrossRef]
- Solassol, J.; Mange, A.; Maudelonde, T. FKBP family proteins as promising new biomarkers for cancer. Curr. Opin. Pharmacol. 2011, 11, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Cheung-Flynn, J.; Prapapanich, V.; Cox, M.B.; Riggs, D.L.; Suarez-Quian, C.; Smith, D.F. Physiological role for the cochaperone FKBP52 in androgen receptor signaling. Mol. Endocrinol. 2005, 19, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Riggs, D.L.; Roberts, P.J.; Chirillo, S.C.; Cheung-Flynn, J.; Prapapanich, V.; Ratajczak, T.; Gaber, R.; Picard, D.; Smith, D.F. The Hsp90-binding peptidylprolyl isomerase FKBP52 potentiates glucocorticoid signaling in vivo. EMBO J. 2003, 22, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wolf, I.M.; Chen, H.; Periyasamy, S.; Chen, Z.; Yong, W.; Shi, S.; Zhao, W.; Xu, J.; Srivastava, A.; et al. FK506-binding protein 52 is essential to uterine reproductive physiology controlled by the progesterone receptor A isoform. Mol. Endocrinol. 2006, 20, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Habara, M.; Kawaguchi, M.; Matsumoto, H.; Hanaki, S.; Masaki, T.; Sato, Y.; Matsuyama, H.; Kunieda, K.; Nakagawa, H.; et al. FKBP51 and FKBP52 regulate androgen receptor dimerization and proliferation in prostate cancer cells. Mol. Oncol. 2022, 16, 940–956. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; D’Angelillo, A.; Pacelli, R.; Staibano, S.; De Luna, E.; Bisogni, R.; Eskelinen, E.L.; Mascolo, M.; Cali, G.; Arra, C.; et al. Role of FK506-binding protein 51 in the control of apoptosis of irradiated melanoma cells. Cell Death Differ. 2010, 17, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D.; Mohammed, Z.; Bacani, J.T.; Lai, R.; Ingham, R.J. The heat shock protein-90 co-chaperone, Cyclophilin 40, promotes ALK-positive, anaplastic large cell lymphoma viability and its expression is regulated by the NPM-ALK oncoprotein. BMC Cancer 2012, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, S.S. An overview of cyclophilins in human cancers. J. Int. Med. Res. 2010, 38, 1561–1574. [Google Scholar] [CrossRef]
- Lu, Z.; Hunter, T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014, 24, 1033–1049. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser-Pornillos, B.K.; Yeager, M. Atomic-level modelling of the HIV capsid. Nature 2011, 469, 424–427. [Google Scholar] [CrossRef]
- Toccafondi, E.; Lener, D.; Negroni, M. HIV-1 Capsid Core: A Bullet to the Heart of the Target Cell. Front. Microbiol. 2021, 12, 652486. [Google Scholar] [CrossRef]
- Luban, J.; Bossolt, K.L.; Franke, E.K.; Kalpana, G.V.; Goff, S.P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 1993, 73, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Franke, E.K.; Yuan, H.E.; Luban, J. Specific incorporation of cyclophilin A into HIV-1 virions. Nature 1994, 372, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Sokolskaja, E.; Sayah, D.M.; Luban, J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 2004, 78, 12800–12808. [Google Scholar] [CrossRef] [PubMed]
- Thali, M.; Bukovsky, A.; Kondo, E.; Rosenwirth, B.; Walsh, C.T.; Sodroski, J.; Gottlinger, H.G. Functional association of cyclophilin A with HIV-1 virions. Nature 1994, 372, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Myszka, D.G.; Yeh, C.; McMurray, M.; Hill, C.P.; Sundquist, W.I. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 1997, 269, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; Strambio-De-Castillia, C.; Campbell, E.M.; Luban, J. Cyclophilin A protects HIV-1 from restriction by human TRIM5alpha. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef]
- Misumi, S.; Inoue, M.; Dochi, T.; Kishimoto, N.; Hasegawa, N.; Takamune, N.; Shoji, S. Uncoating of human immunodeficiency virus type 1 requires prolyl isomerase Pin1. J. Biol. Chem. 2010, 285, 25185–25195. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Khan, M.; Yedavalli, V.R.; Yeung, M.L.; Strebel, K.; Jeang, K.T. Human immunodeficiency virus type 1 replication and regulation of APOBEC3G by peptidyl prolyl isomerase Pin1. J. Virol. 2008, 82, 9928–9936. [Google Scholar] [CrossRef] [PubMed]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Manganaro, L.; Lusic, M.; Gutierrez, M.I.; Cereseto, A.; Del Sal, G.; Giacca, M. Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat. Med. 2010, 16, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, X.; Zhu, Y.; Shen, Y.; Wang, Y.; Lu, P.; Jiang, Z.; Pan, H.; Yang, J.; Xun, J.; et al. FKBP3 Induces Human Immunodeficiency Virus Type 1 Latency by Recruiting Histone Deacetylase 1/2 to the Viral Long Terminal Repeat. mBio 2021, 12, e0079521. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Shimotohno, K. The roles of hepatitis C virus proteins in modulation of cellular functions: A novel action mechanism of the HCV core protein on gene regulation by nuclear hormone receptors. Cancer Sci. 2003, 94, 937–943. [Google Scholar] [CrossRef]
- Nakagawa, M.; Sakamoto, N.; Enomoto, N.; Tanabe, Y.; Kanazawa, N.; Koyama, T.; Kurosaki, M.; Maekawa, S.; Yamashiro, T.; Chen, C.H.; et al. Specific inhibition of hepatitis C virus replication by cyclosporin A. Biochem. Biophys. Res. Commun. 2004, 313, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Ishii, N.; Hijikata, M.; Inoue, D.; Murata, T.; Miyanari, Y.; Shimotohno, K. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell. 2005, 19, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Bobardt, M.D.; Lim, P.; Gallay, P.A. Cyclophilin A-independent recruitment of NS5A and NS5B into hepatitis C virus replication complexes. J. Gen. Virol. 2010, 91 Pt 5, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.S.; Hwang, S.B. Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type III alpha and regulates viral propagation. J. Biol. Chem. 2011, 286, 11290–11298. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Utsunomiya, T.; Wakiyama, S.; Hashimoto, M.; Fukuzawa, K.; Ezaki, T.; Hanai, T.; Inoue, H.; Mori, M. Specific gene-expression profiles of noncancerous liver tissue predict the risk for multicentric occurrence of hepatocellular carcinoma in hepatitis C virus-positive patients. Ann. Surg. Oncol. 2006, 13, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Nishi, M.; Miyakawa, K.; Matsunaga, S.; Khatun, H.; Yamaoka, Y.; Watashi, K.; Sugiyama, M.; Kimura, H.; Wakita, T.; Ryo, A. Prolyl Isomerase Pin1 Regulates the Stability of Hepatitis B Virus Core Protein. Front. Cell Dev. Biol. 2020, 8, 26. [Google Scholar] [CrossRef]
- Pang, R.; Lee, T.K.; Poon, R.T.; Fan, S.T.; Wong, K.B.; Kwong, Y.L.; Tse, E. Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis. Gastroenterology 2007, 132, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Banerjee, A.; Chandra, P.K.; Biswas, A.; Panigrahi, R.; Mahapatra, P.K.; Panda, C.K.; Chakrabarti, S.; Bhattacharya, S.K.; Chakravarty, R. Analysis of hepatitis B virus X gene phylogeny, genetic variability and its impact on pathogenesis: Implications in Eastern Indian HBV carriers. Virology 2008, 382, 190–198. [Google Scholar] [CrossRef]
- Saeed, U.; Piracha, Z.Z.; Kwon, H.; Kim, J.; Kalsoom, F.; Chwae, Y.J.; Park, S.; Shin, H.J.; Lee, H.W.; Lim, J.H.; et al. The HBV Core Protein and Core Particle Both Bind to the PPiase Par14 and Par17 to Enhance Their Stabilities and HBV Replication. Front. Microbiol. 2021, 12, 795047. [Google Scholar] [CrossRef]
- Foster, R.T.; Ure, D.R.; Trepanier, D.J.; Gallay, P. The cyclophilin inhibitor CRV431 prevents both cyclophilin A-HBx complex formation and HBV replication. J. Hepatol. 2017, 66, S699. [Google Scholar] [CrossRef]
- Tian, X.; Zhao, C.; Zhu, H.; She, W.; Zhang, J.; Liu, J.; Li, L.; Zheng, S.; Wen, Y.M.; Xie, Y. Hepatitis B virus (HBV) surface antigen interacts with and promotes cyclophilin a secretion: Possible link to pathogenesis of HBV infection. J. Virol. 2010, 84, 3373–3381. [Google Scholar] [CrossRef]
- Gallay, P.; Ure, D.; Bobardt, M.; Chatterji, U.; Ou, J.; Trepanier, D.; Foster, R. The cyclophilin inhibitor CRV431 inhibits liver HBV DNA and HBsAg in transgenic mice. PLoS ONE 2019, 14, e0217433. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.; Chokshi, S.; Chatterji, U.; Riva, A.; Bobardt, M.; Williams, R.; Gallay, P.; Naoumov, N.V. Alisporivir inhibition of hepatocyte cyclophilins reduces HBV replication and hepatitis B surface antigen production. Gastroenterology 2015, 148, 403–414.e7. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Ariumi, Y.; Nishida, N.; Yamamoto, R.; Bauer, G.; Gojobori, T.; Shimotohno, K.; Mizokami, M. SARS-CoV-2 infections and COVID-19 mortalities strongly correlate with ACE1 I/D genotype. Gene 2020, 758, 144944. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ma, Z.; Zhang, Y.; Zhang, M.; Shi, X.; Zhang, M.; Zhang, W.; Liu, W. The role of cyclophilins in viral infection and the immune response. J. Infect. 2022, 85, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Kanna, M.; Nakatsu, Y.; Yamamotoya, T.; Encinas, J.; Ito, H.; Okabe, T.; Asano, T.; Sakaguchi, T. Roles of peptidyl prolyl isomerase Pin1 in viral propagation. Front. Cell Dev. Biol. 2022, 10, 1005325. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Greenwood, A.; Binder, L.; Bigio, E.H.; Denial, S.; Nicholson, L.; Zhou, X.Z.; Lu, K.P. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell 2012, 149, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.L.; Pastorino, L.; Zhou, X.Z.; Lu, K.P. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3beta (GSK3beta) activity: Novel mechanism for Pin1 to protect against Alzheimer disease. J. Biol. Chem. 2012, 287, 6969–6973. [Google Scholar] [CrossRef]
- Lucia, P.; Asami, K.; Xiao Zhen, Z.; Kun Ping, L. Pin1 Protects Against Alzheimer’s Disease: One Goal, Multiple Mechanisms. In Understanding Alzheimer’s Disease; Inga, Z., Ed.; IntechOpen: Rijeka, Croatia, 2013; p. 6. [Google Scholar]
- Lee, T.H.; Pastorino, L.; Lu, K.P. Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev. Mol. Med. 2011, 13, e21. [Google Scholar] [CrossRef]
- Ryo, A.; Togo, T.; Nakai, T.; Hirai, A.; Nishi, M.; Yamaguchi, A.; Suzuki, K.; Hirayasu, Y.; Kobayashi, H.; Perrem, K.; et al. Prolyl-isomerase Pin1 accumulates in lewy bodies of parkinson disease and facilitates formation of alpha-synuclein inclusions. J. Biol. Chem. 2006, 281, 4117–4125. [Google Scholar] [CrossRef] [PubMed]
- Favretto, F.; Baker, J.D.; Strohaker, T.; Andreas, L.B.; Blair, L.J.; Becker, S.; Zweckstetter, M. The Molecular Basis of the Interaction of Cyclophilin A with alpha-Synuclein. Angew. Chem. Int. Ed. Engl. 2020, 59, 5643–5646. [Google Scholar] [CrossRef] [PubMed]
- Caminati, G.; Martina, M.R.; Menichetti, S.; Procacci, P. Blocking the FKBP12 induced dendrimeric burst in aberrant aggregation of alpha-synuclein by using the ElteN378 synthetic inhibitor. J. Enzym. Inhib. Med. Chem. 2019, 34, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, F.; Govoni, S.; Racchi, M.; Lanni, C. The Peptidyl-prolyl Isomerase Pin1 in Neuronal Signaling: From Neurodevelopment to Neurodegeneration. Mol. Neurobiol. 2021, 58, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Sedrani, R.; Kallen, J.; Martin Cabrejas, L.M.; Papageorgiou, C.D.; Senia, F.; Rohrbach, S.; Wagner, D.; Thai, B.; Jutzi Eme, A.M.; France, J.; et al. Sanglifehrin-cyclophilin interaction: Degradation work, synthetic macrocyclic analogues, X-ray crystal structure, and binding data. J. Am. Chem. Soc. 2003, 125, 3849–3859. [Google Scholar] [CrossRef]
- Ptak, R.G.; Gallay, P.A.; Jochmans, D.; Halestrap, A.P.; Ruegg, U.T.; Pallansch, L.A.; Bobardt, M.D.; de Bethune, M.P.; Neyts, J.; De Clercq, E.; et al. Inhibition of human immunodeficiency virus type 1 replication in human cells by Debio-025, a novel cyclophilin binding agent. Antimicrob. Agents Chemother. 2008, 52, 1302–1317. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Kaul, A.; De Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef]
- Ma, S.; Boerner, J.E.; TiongYip, C.; Weidmann, B.; Ryder, N.S.; Cooreman, M.P.; Lin, K. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob. Agents Chemother. 2006, 50, 2976–2982. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.; Serrano, S.S.; Gronberg, A.; Massoumi, R.; Hansson, M.J.; Gallay, P. Cyclophilin Inhibitor NV556 Reduces Fibrosis and Hepatocellular Carcinoma Development in Mice With Non-Alcoholic Steatohepatitis. Front. Pharmacol. 2019, 10, 1129. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; Scorneaux, B.; Huang, Z.; Murray, M.G.; Wring, S.; Smitley, C.; Harris, R.; Erdmann, F.; Fischer, G.; Ribeill, Y. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 2010, 54, 660–672. [Google Scholar] [CrossRef]
- Serrano, S.S.; Tavecchio, M.; Gronberg, A.; Sime, W.; Jemaa, M.; Moss, S.; Gregory, M.A.; Gallay, P.; Elmer, E.; Hansson, M.J.; et al. Novel Cyclophilin Inhibitor Decreases Cell Proliferation and Tumor Growth in Models of Hepatocellular Carcinoma. Cancers 2021, 13, 3041. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.S.; Gronberg, A.; Longato, L.; Rombouts, K.; Kuo, J.; Gregory, M.; Moss, S.; Elmer, E.; Mazza, G.; Gallay, P.; et al. Evaluation of NV556, a Novel Cyclophilin Inhibitor, as a Potential Antifibrotic Compound for Liver Fibrosis. Cells 2019, 8, 1409. [Google Scholar] [CrossRef] [PubMed]
- Gaali, S.; Kirschner, A.; Cuboni, S.; Hartmann, J.; Kozany, C.; Balsevich, G.; Namendorf, C.; Fernandez-Vizarra, P.; Sippel, C.; Zannas, A.S.; et al. Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat. Chem. Biol. 2015, 11, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.B.; Zou, C.L.; Duan, Y.X.; Wu, F.; Li, G. Activity guided isolation and modification of juglone from Juglans regia as potent cytotoxic agent against lung cancer cell lines. BMC Complement. Altern. Med. 2015, 15, 396. [Google Scholar] [CrossRef]
- Campaner, E.; Rustighi, A.; Zannini, A.; Cristiani, A.; Piazza, S.; Ciani, Y.; Kalid, O.; Golan, G.; Baloglu, E.; Shacham, S.; et al. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun. 2017, 8, 15772. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Pinch, B.J.; Doctor, Z.M.; Nabet, B.; Browne, C.M.; Seo, H.S.; Mohardt, M.L.; Kozono, S.; Lian, X.; Manz, T.D.; Chun, Y.; et al. Identification of a potent and selective covalent Pin1 inhibitor. Nat. Chem. Biol. 2020, 16, 979–987. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, R.; Li, J.; Pei, Y.; Wang, S.; Xu, P.; Wang, M.; Wen, Y.; Zhang, H.; Du, D.; et al. Computational and Structure-Based Development of High Potent Cell-Active Covalent Inhibitor Targeting the Peptidyl-Prolyl Isomerase NIMA-Interacting-1 (Pin1). J. Med. Chem. 2022, 65, 2174–2190. [Google Scholar] [CrossRef]
- Dubiella, C.; Pinch, B.J.; Koikawa, K.; Zaidman, D.; Poon, E.; Manz, T.D.; Nabet, B.; He, S.; Resnick, E.; Rogel, A.; et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat. Chem. Biol. 2021, 17, 954–963. [Google Scholar] [CrossRef]
- Guo, C.; Hou, X.; Dong, L.; Dagostino, E.; Greasley, S.; Ferre, R.; Marakovits, J.; Johnson, M.C.; Matthews, D.; Mroczkowski, B.; et al. Structure-based design of novel human Pin1 inhibitors (I). Bioorg. Med. Chem. Lett. 2009, 19, 5613–5616. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Marakovits, J.; Hou, X.; Guo, C.; Greasley, S.; Dagostino, E.; Ferre, R.; Johnson, M.C.; Kraynov, E.; Thomson, J.; et al. Structure-based design of novel human Pin1 inhibitors (II). Bioorg. Med. Chem. Lett. 2010, 20, 2210–2214. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Hou, X.; Dong, L.; Marakovits, J.; Greasley, S.; Dagostino, E.; Ferre, R.; Johnson, M.C.; Humphries, P.S.; Li, H.; et al. Structure-based design of novel human Pin1 inhibitors (III): Optimizing affinity beyond the phosphate recognition pocket. Bioorg. Med. Chem. Lett. 2014, 24, 4187–4191. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Di Stefano, M.; Estevez, J.A.; Minutolo, F.; Granchi, C.; Giordano, A.; Parisi, S.; Mauceri, M.; Canzonieri, V.; Macchia, M.; et al. New PIN1 inhibitors identified through a pharmacophore-driven, hierarchical consensus docking strategy. J. Enzym. Inhib. Med. Chem. 2022, 37, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Zenke, G.; Strittmatter, U.; Fuchs, S.; Quesniaux, V.F.; Brinkmann, V.; Schuler, W.; Zurini, M.; Enz, A.; Billich, A.; Sanglier, J.J.; et al. Sanglifehrin A, a novel cyclophilin-binding compound showing immunosuppressive activity with a new mechanism of action. J. Immunol. 2001, 166, 7165–7171. [Google Scholar] [CrossRef]
- Borel, J.F.; Feurer, C.; Gubler, H.U.; Stähelin, H. Biological effects of cyclosporin A: A new antilymphocytic agent. Agents Actions 1976, 6, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.M.; Miller, J.F.; Shevach, E.M. Mechanism of action of cyclosporine A in vivo. II. T cell priming in vivo to alloantigen can be mediated by an IL-2-independent cyclosporine A-resistant pathway. J. Immunol. 1990, 144, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.; Remberger, M.; Hassan, Z.; Omazic, B.; Mattsson, J.; Ringden, O. GVHD prophylaxis using low-dose cyclosporine improves survival in leukaemic recipients of HLA-identical sibling transplants. Eur. J. Haematol. 2010, 84, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Flores, R.; Bojalil, R.; Benitez, J.C.; Ledesma-Soto, Y.; Terrazas, C.A.; Rodriguez-Sosa, M.; Terrazas, L.I. Consecutive low doses of cyclosporine A induce pro-inflammatory cytokines and accelerate allograft skin rejection. Molecules 2011, 16, 3969–3984. [Google Scholar] [CrossRef]
- Flores, C.; Fouquet, G.; Moura, I.C.; Maciel, T.T.; Hermine, O. Lessons to Learn From Low-Dose Cyclosporin-A: A New Approach for Unexpected Clinical Applications. Front. Immunol. 2019, 10, 588. [Google Scholar] [CrossRef]
- Ross, H.J.; Cho, J.; Osann, K.; Wong, S.F.; Ramsinghani, N.; Williams, J.; Downey-Hurtado, N.; Slater, L.M. Phase I/II trial of low dose cyclosporin A with EP for advanced non-small cell lung cancer. Lung Cancer 1997, 18, 189–198. [Google Scholar] [CrossRef]
- Nicolas, D.; Ambrosioni, J.; Sued, O.; Brunet, M.; Lopez-Dieguez, M.; Manzardo, C.; Aguero, F.; Tuset, M.; Plana, M.; Guardo, A.C.; et al. Cyclosporine A in addition to standard ART during primary HIV-1 infection: Pilot randomized clinical trial. J. Antimicrob. Chemother. 2017, 72, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Kaptein, S.; Paeshuyse, J.; Vliegen, I.; Dumont, J.M.; Vuagniaux, G.; Neyts, J. Debio 025, a cyclophilin binding molecule, is highly efficient in clearing hepatitis C virus (HCV) replicon-containing cells when used alone or in combination with specifically targeted antiviral therapy for HCV (STAT-C) inhibitors. Antimicrob. Agents Chemother. 2009, 53, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Horban, A.; Gallay, P.; Bobardt, M.; Selvarajah, S.; Wiercinska-Drapalo, A.; Siwak, E.; Cielniak, I.; Higersberger, J.; Kierkus, J.; et al. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 2008, 47, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Feinman, S.V.; Jablkowski, M.; Horban, A.; Kryczka, W.; Pawlowska, M.; Heathcote, J.E.; Mazzella, G.; Vandelli, C.; Nicolas-Metral, V.; et al. The cyclophilin inhibitor Debio 025 combined with PEG IFN alpha 2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 2009, 49, 1460–1468. [Google Scholar] [CrossRef] [PubMed]
- Rosenwirth, B.; Billich, A.; Datema, R.; Donatsch, P.; Hammerschmid, F.; Harrison, R.; Hiestand, P.; Jaksche, H.; Mayer, P.; Peichl, P.; et al. Inhibition of human immunodeficiency virus type 1 replication by SDZ NIM 811, a nonimmunosuppressive cyclosporine analog. Antimicrob. Agents Chemother. 1994, 38, 1763–1772. [Google Scholar] [CrossRef]
- Lawitz, E.; Godofsky, E.; Rouzier, R.; Marbury, T.; Nguyen, T.; Ke, J.; Huang, M.; Praestgaard, J.; Serra, D.; Evans, T.G. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antivir. Res. 2011, 89, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.J.; Mattiasson, G.; Mansson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.M.; Besseghir, K.; Elmer, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef]
- Gallay, P.A.; Bobardt, M.D.; Chatterji, U.; Trepanier, D.J.; Ure, D.; Ordonez, C.; Foster, R. The Novel Cyclophilin Inhibitor CPI-431-32 Concurrently Blocks HCV and HIV-1 Infections via a Similar Mechanism of Action. PLoS ONE 2015, 10, e0134707. [Google Scholar] [CrossRef] [PubMed]
- Sanglier, J.J.; Quesniaux, V.; Fehr, T.; Hofmann, H.; Mahnke, M.; Memmert, K.; Schuler, W.; Zenke, G.; Gschwind, L.; Maurer, C.; et al. Sanglifehrins A, B, C and D, novel cyclophilin-binding compounds isolated from Streptomyces sp. A92-308110 I. Taxonomy, fermentation, isolation and biological activity. J. Antibiot. 1999, 52, 466–473. [Google Scholar] [CrossRef]
- Zhang, L.H.; Liu, J.O. Sanglifehrin A, a novel cyclophilin-binding immunosuppressant, inhibits IL-2-dependent T cell proliferation at the G1 phase of the cell cycle. J. Immunol. 2001, 166, 5611–5618. [Google Scholar] [CrossRef]
- Pua, K.H.; Stiles, D.T.; Sowa, M.E.; Verdine, G.L. IMPDH2 Is an Intracellular Target of the Cyclophilin A and Sanglifehrin A Complex. Cell Rep. 2017, 18, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Bobardt, M.; Obeid, S.; Chatterji, U.; Coates, N.J.; Foster, T.; Gallay, P.; Leyssen, P.; Moss, S.J.; Neyts, J.; et al. Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob. Agents Chemother. 2011, 55, 1975–1981. [Google Scholar] [CrossRef]
- Moss, S.J.; Bobardt, M.; Leyssen, P.; Coates, N.; Chatterji, U.; Dejian, X.; Foster, T.; Liu, J.; Nur-e-Alam, M.; Suthar, D.; et al. Sangamides, a new class of cyclophilin-inhibiting host-targeted antivirals for treatment of HCV infection. MedChemComm 2012, 3, 944–949. [Google Scholar] [CrossRef]
- Immecke, S.N.; Baal, N.; Wilhelm, J.; Bechtel, J.; Knoche, A.; Bein, G.; Hackstein, H. The cyclophilin-binding agent Sanglifehrin A is a dendritic cell chemokine and migration inhibitor. PLoS ONE 2011, 6, e18406. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, H.A.; Chrysovergi, M.A.; Han, H.; Kabir, F.; Lister, R.T.; Chang, C.F.; Black, K.E.; Lagares, D.; Woo, C.M. Sanglifehrin A mitigates multi-organ fibrosis in vivo by inducing secretion of the collagen chaperone cyclophilin B. bioRxiv 2023. [Google Scholar] [CrossRef]
- Bobardt, M.; Hansson, M.J.; Mayo, P.; Ure, D.; Foster, R.; Gallay, P. Structurally distinct cyclosporin and sanglifehrin analogs CRV431 and NV556 suppress established HCV infection in humanized-liver mice. PLoS ONE 2020, 15, e0237236. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.S.; Tavecchio, M.; Mallik, J.; Gronberg, A.; Elmer, E.; Kifagi, C.; Gallay, P.; Hansson, M.J.; Massoumi, R. Synergistic Effects of Sanglifehrin-Based Cyclophilin Inhibitor NV651 with Cisplatin in Hepatocellular Carcinoma. Cancers 2022, 14, 4553. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hatanaka, H.; Hashimoto, M.; Nishiyama, M.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; Imanaka, H. FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics. J. Antibiot. 1987, 40, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Hatanaka, H.; Miyata, S.; Inamura, N.; Nishiyama, M.; Yajima, T.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J. Antibiot. 1987, 40, 1256–1265. [Google Scholar] [CrossRef]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef]
- Sehgal, S.N.; Baker, H.; Vezina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Vanduyne, G.D.; Standaert, R.F.; Schreiber, S.L.; Clardy, J. Atomic-Structure of the Rapamycin Human Immunophilin Fkbp-12 Complex. J. Am. Chem. Soc. 1991, 113, 7433–7434. [Google Scholar] [CrossRef]
- Vanduyne, G.D.; Standaert, R.F.; Karplus, P.A.; Schreiber, S.L.; Clardy, J. Atomic Structures of the Human Immunophilin Fkbp-12 Complexes with Fk506 and Rapamycin. J. Mol. Biol. 1993, 229, 105–124. [Google Scholar] [CrossRef]
- Bocquet, A.; Lorent, G.; Fuks, B.; Grimee, R.; Talaga, P.; Daliers, J.; Klitgaard, H. Failure of GPI compounds to display neurotrophic activity in vitro and in vivo. Eur. J. Pharmacol. 2001, 415, 173–180. [Google Scholar] [CrossRef]
- Eberling, J.L.; Pivirotto, P.; Bringas, J.; Steiner, J.P.; Kordower, J.H.; Chu, Y.; Emborg, M.E.; Bankiewicz, K.S. The immunophilin ligand GPI-1046 does not have neuroregenerative effects in MPTP-treated monkeys. Exp. Neurol. 2002, 178, 236–242. [Google Scholar] [CrossRef]
- Odom, A.; Del Poeta, M.; Perfect, J.; Heitman, J. The immunosuppressant FK506 and its nonimmunosuppressive analog L-685,818 are toxic to Cryptococcus neoformans by inhibition of a common target protein. Antimicrob. Agents Chemother. 1997, 41, 156–161. [Google Scholar] [CrossRef]
- Cruz, M.C.; Del Poeta, M.; Wang, P.; Wenger, R.; Zenke, G.; Quesniaux, V.F.; Movva, N.R.; Perfect, J.R.; Cardenas, M.E.; Heitman, J. Immunosuppressive and nonimmunosuppressive cyclosporine analogs are toxic to the opportunistic fungal pathogen Cryptococcus neoformans via cyclophilin-dependent inhibition of calcineurin. Antimicrob. Agents Chemother. 2000, 44, 143–149. [Google Scholar] [CrossRef]
- Steinbach, W.J.; Cramer, R.A., Jr.; Perfect, B.Z.; Asfaw, Y.G.; Sauer, T.C.; Najvar, L.K.; Kirkpatrick, W.R.; Patterson, T.F.; Benjamin, D.K., Jr.; Heitman, J.; et al. Calcineurin controls growth, morphology, and pathogenicity in Aspergillus fumigatus. Eukaryot. Cell 2006, 5, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Norville, I.H.; O’Shea, K.; Sarkar-Tyson, M.; Zheng, S.; Titball, R.W.; Varani, G.; Harmer, N.J. The structure of a Burkholderia pseudomallei immunophilin-inhibitor complex reveals new approaches to antimicrobial development. Biochem. J. 2011, 437, 413–422. [Google Scholar] [CrossRef]
- Rasch, J.; Theuerkorn, M.; Unal, C.; Heinsohn, N.; Tran, S.; Fischer, G.; Weiwad, M.; Steinert, M. Novel Cycloheximide Derivatives Targeting the Moonlighting Protein Mip Exhibit Specific Antimicrobial Activity Against Legionella pneumophila. Front. Bioeng. Biotechnol. 2015, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- De Leon, J.T.; Iwai, A.; Feau, C.; Garcia, Y.; Balsiger, H.A.; Storer, C.L.; Suro, R.M.; Garza, K.M.; Lee, S.; Kim, Y.S.; et al. Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11878–11883. [Google Scholar] [CrossRef] [PubMed]
- D’Arrigo, P.; Russo, M.; Rea, A.; Tufano, M.; Guadagno, E.; Del Basso De Caro, M.L.; Pacelli, R.; Hausch, F.; Staibano, S.; Ilardi, G.; et al. A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget 2017, 8, 68291–68304. [Google Scholar] [CrossRef] [PubMed]
- Annett, S.; Moore, G.; Short, A.; Marshall, A.; McCrudden, C.; Yakkundi, A.; Das, S.; McCluggage, W.G.; Nelson, L.; Harley, I.; et al. FKBPL-based peptide, ALM201, targets angiogenesis and cancer stem cells in ovarian cancer. Br. J. Cancer. 2020, 122, 361–371. [Google Scholar] [CrossRef]
- Hennig, L.; Christner, C.; Kipping, M.; Schelbert, B.; Rucknagel, K.P.; Grabley, S.; Kullertz, G.; Fischer, G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 1998, 37, 5953–5960. [Google Scholar] [CrossRef]
- Chao, S.H.; Greenleaf, A.L.; Price, D.H. Juglone, an inhibitor of the peptidyl-prolyl isomerase Pin1, also directly blocks transcription. Nucleic Acids Res. 2001, 29, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Pavan, V.; Ribaudo, G.; Zorzan, M.; Redaelli, M.; Pezzani, R.; Mucignat-Caretta, C.; Zagotto, G. Antiproliferative activity of Juglone derivatives on rat glioma. Nat. Prod. Res. 2017, 31, 632–638. [Google Scholar] [CrossRef]
- Wang, J.; Liu, K.; Wang, X.F.; Sun, D.J. Juglone reduces growth and migration of U251 glioblastoma cells and disrupts angiogenesis. Oncol. Rep. 2017, 38, 1959–1966. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, H.; Xu, Y.; Zhang, J.; Zhu, W.; Zhang, Y.; Chen, L.; Hua, W.; Mao, Y. Juglone induces apoptosis of tumor stem-like cells through ROS-p38 pathway in glioblastoma. BMC Neurol. 2017, 17, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhu, W.; Zhang, R.; Zhang, M.; Zhao, J.; Hou, J.; Zhang, W. Targeted juglone blocks the invasion and metastasis of HPV-positive cervical cancer cells. J. Pharmacol. Sci. 2019, 140, 211–217. [Google Scholar] [CrossRef]
- Avci, E.; Arikoglu, H.; Erkoc Kaya, D. Investigation of juglone effects on metastasis and angiogenesis in pancreatic cancer cells. Gene 2016, 588, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhang, Y.; Zhang, Z.; Che, D.; Lv, H. Juglone loaded poloxamer 188/phospholipid mixed micelles evaluated in vitro and in vivo in breast cancer. Int. J. Pharm. 2016, 515, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Sajadimajd, S.; Yazdanparast, R. Sensitizing effect of juglone is mediated by down regulation of Notch1 signaling pathway in trastuzumab-resistant SKBR3 cells. Apoptosis 2017, 22, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Song, X.; Zhao, J.; Zhu, W.; Hou, J.; Wang, Y.; Zhang, W. Juglone Inhibits Proliferation of HPV-Positive Cervical Cancer Cells Specifically. Biol. Pharm. Bull. 2019, 42, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, R.; Kushiyama, A.; Seno, Y.; Nakatsu, Y.; Matsunaga, Y.; Fukushima, T.; Tsuchiya, Y.; Sakoda, H.; Fujishiro, M.; Yamamotoya, T.; et al. Pin1 Inhibitor Juglone Exerts Anti-Oncogenic Effects on LNCaP and DU145 Cells despite the Patterns of Gene Regulation by Pin1 Differing between These Cell Lines. PLoS ONE 2015, 10, e0127467. [Google Scholar] [CrossRef]
- Fang, F.; Chen, S.; Ma, J.; Cui, J.; Li, Q.; Meng, G.; Wang, L. Juglone suppresses epithelial-mesenchymal transition in prostate cancer cells via the protein kinase B/glycogen synthase kinase-3beta/Snail signaling pathway. Oncol. Lett. 2018, 16, 2579–2584. [Google Scholar]
- Meskelevicius, D.; Sidlauskas, K.; Bagdonaviciute, R.; Liobikas, J.; Majiene, D. Juglone Exerts Cytotoxic, Anti-proliferative and Anti-invasive Effects on Glioblastoma Multiforme in a Cell Culture Model. Anticancer Agents Med. Chem. 2016, 16, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Gokturk, F.; Erkoc-Kaya, D.; Arikoglu, H. Juglone can inhibit angiogenesis and metastasis in pancreatic cancer cells by targeting Wnt/beta-catenin signaling. Bratisl. Med. J. Bratisl. Lek. Listy 2021, 122, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Kviecinski, M.R.; Pedrosa, R.C.; Felipe, K.B.; Farias, M.S.; Glorieux, C.; Valenzuela, M.; Sid, B.; Benites, J.; Valderrama, J.A.; Verrax, J.; et al. Inhibition of cell proliferation and migration by oxidative stress from ascorbate-driven juglone redox cycling in human bladder-derived T24 cells. Biochem. Biophys. Res. Commun. 2012, 421, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Bayram, D.; Armagan, I.; Ozgocmen, M.; Senol, N.; Calapoglu, M. Determination of Apoptotic Effect of Juglone on Human Bladder Cancer TCC-SUP and RT-4 Cells: An In Vitro Study. J. Environ. Pathol. Toxicol. Oncol. 2018, 37, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Sun, R.; Kim, J. Juglone and KPT6566 Suppress the Tumorigenic Potential of CD44(+)CD133(+) Tumor-Initiating Caco-2 Cells In Vitro and In Vivo. Front. Cell Dev. Biol. 2022, 10, 861045. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.T.; Li, Y.; Chu, P.; Ma, X.D.; Tang, Z.Y.; Sun, Z.L. Molecular biological mechanism of action in cancer therapies: Juglone and its derivatives, the future of development. Biomed. Pharmacother. 2022, 148, 112785. [Google Scholar] [CrossRef]
- Wildemann, D.; Erdmann, F.; Alvarez, B.H.; Stoller, G.; Zhou, X.Z.; Fanghanel, J.; Schutkowski, M.; Lu, K.P.; Fischer, G. Nanomolar inhibitors of the peptidyl prolyl cis/trans isomerase Pin1 from combinatorial peptide libraries. J. Med. Chem. 2006, 49, 2147–2150. [Google Scholar] [CrossRef] [PubMed]
- Daum, S.; Erdmann, F.; Fischer, G.; Feaux de Lacroix, B.; Hessamian-Alinejad, A.; Houben, S.; Frank, W.; Braun, M. Aryl indanyl ketones: Efficient inhibitors of the human peptidyl prolyl cis/trans isomerase Pin1. Angew. Chem. Int. Ed. Engl. 2006, 45, 7454–7458. [Google Scholar] [CrossRef] [PubMed]
- Subedi, A.; Shimizu, T.; Ryo, A.; Sanada, E.; Watanabe, N.; Osada, H. Discovery of novel selenium derivatives as Pin1 inhibitors by high-throughput screening. Biochem. Biophys. Res. Commun. 2016, 474, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, L.; Wang, S.; Li, X.; Ma, T.; Liu, D.; Jing, Y.; Zhao, L. Design and synthesis of novel 2-substituted 11-keto-boswellic acid heterocyclic derivatives as anti-prostate cancer agents with Pin1 inhibition ability. Eur. J. Med. Chem. 2017, 126, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Jin, J.; Chen, H.; Cao, R.; Chen, X.; Xu, B. Synthesis and biological evaluation of pyrimidine derivatives as novel human Pin1 inhibitors. Bioorg. Med. Chem. 2018, 26, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Wang, X.; Cui, G.; Xu, B. Design, synthesis and biological evaluation of novel thiazole-based derivatives as human Pin1 inhibitors. Bioorg. Med. Chem. 2021, 29, 115878. [Google Scholar] [CrossRef] [PubMed]
- Sutton, L.M.; Warmuth, M.A.; Petros, W.P.; Winer, E.P. Pharmacokinetics and clinical impact of all-trans retinoic acid in metastatic breast cancer: A phase II trial. Cancer Chemother. Pharmacol. 1997, 40, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Gonzalez-De la Rosa, C.H.; Arechaga-Ocampo, E.; Villanueva-Rodriguez, G.; Ceron-Lizarraga, T.L.; Martinez-Barrera, L.; Vazquez-Manriquez, M.E.; Rios-Trejo, M.A.; Alvarez-Avitia, M.A.; Hernandez-Pedro, N.; et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3463–3471. [Google Scholar] [CrossRef]
- Kocher, H.M.; Basu, B.; Froeling, F.E.M.; Sarker, D.; Slater, S.; Carlin, D.; de Souza, N.M.; De Paepe, K.N.; Goulart, M.R.; Hughes, C.; et al. Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nat. Commun. 2020, 11, 4841. [Google Scholar] [CrossRef] [PubMed]
- Hanna, G.J.; ONeill, A.; Cutler, J.M.; Flynn, M.; Vijaykumar, T.; Clark, J.R.; Wirth, L.J.; Lorch, J.H.; Park, J.C.; Mito, J.K.; et al. A phase II trial of all-trans retinoic acid (ATRA) in advanced adenoid cystic carcinoma. Oral Oncol. 2021, 119, 105366. [Google Scholar] [CrossRef] [PubMed]
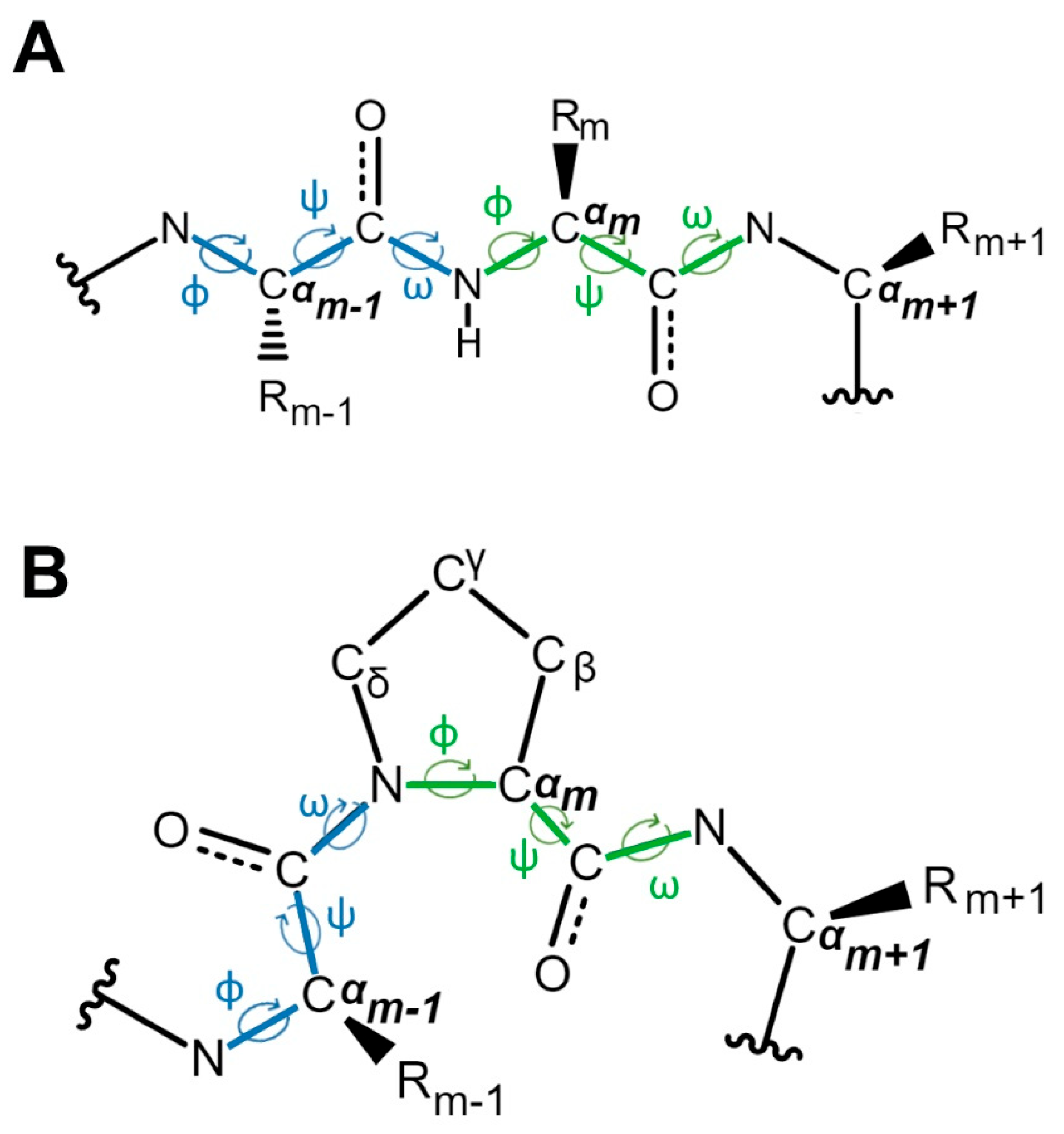
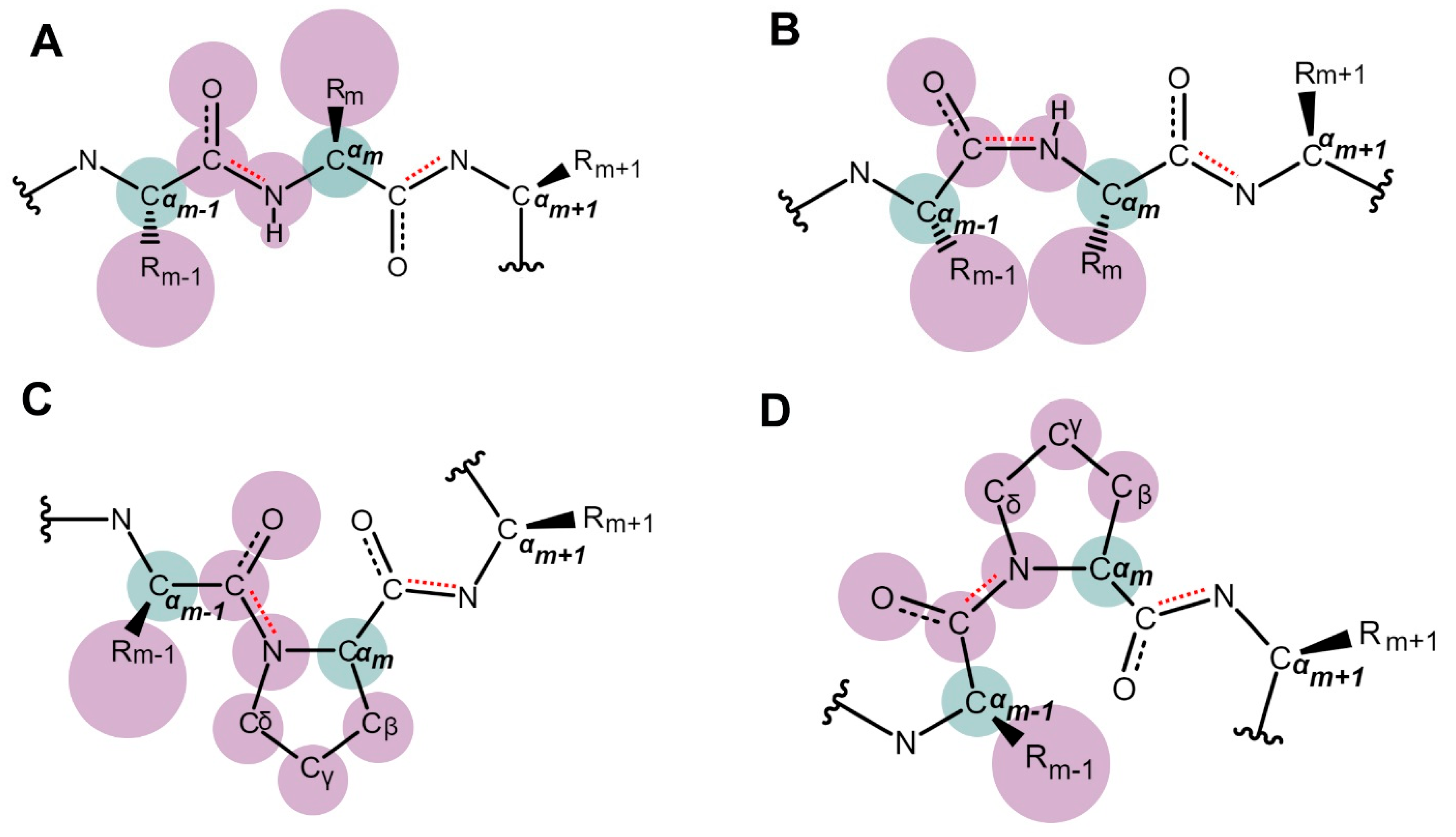
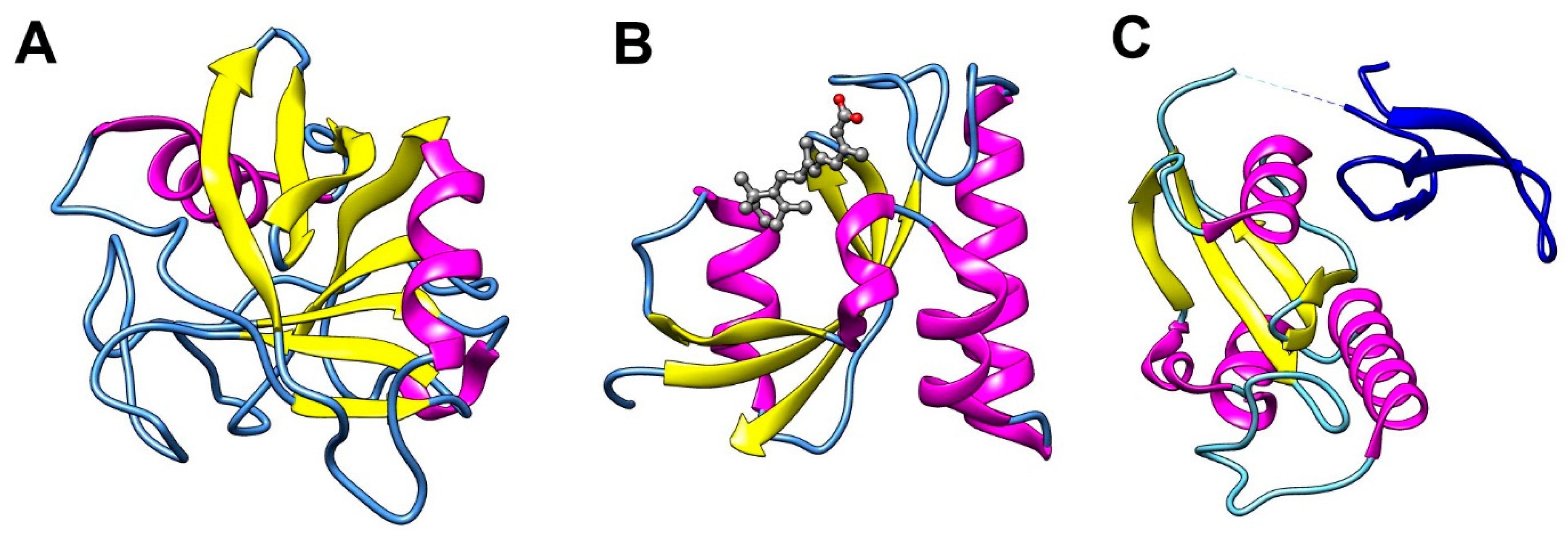

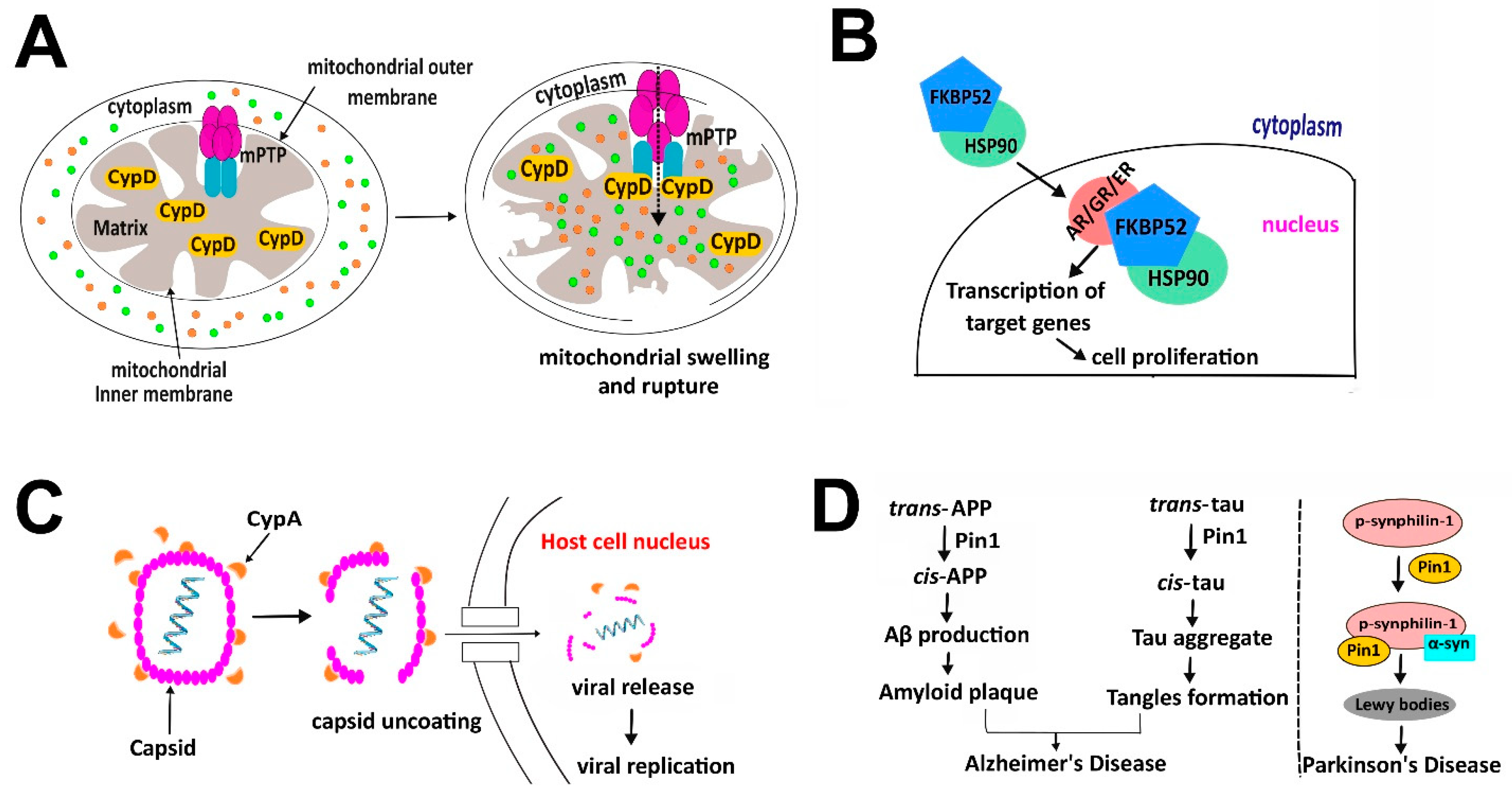
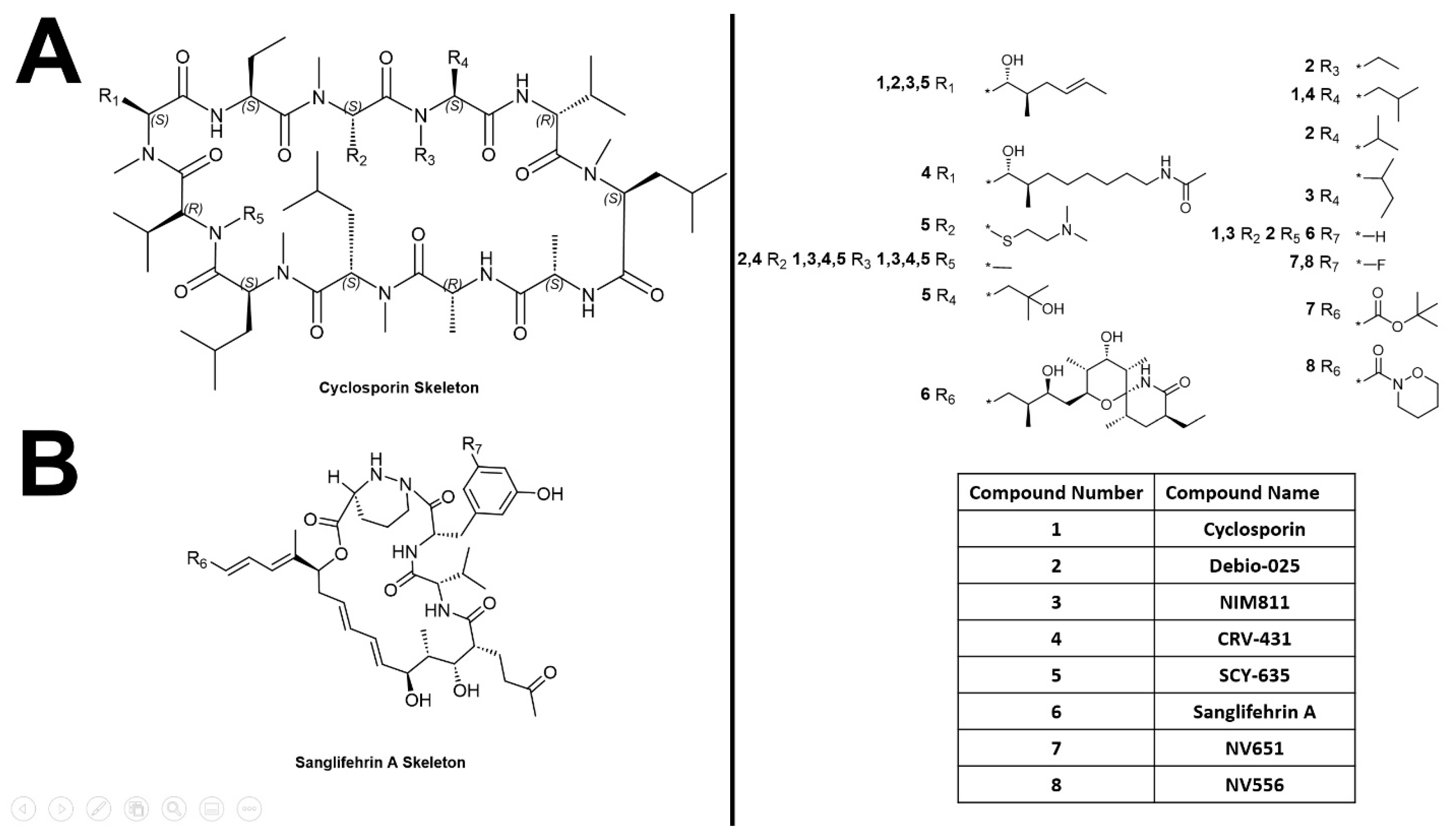
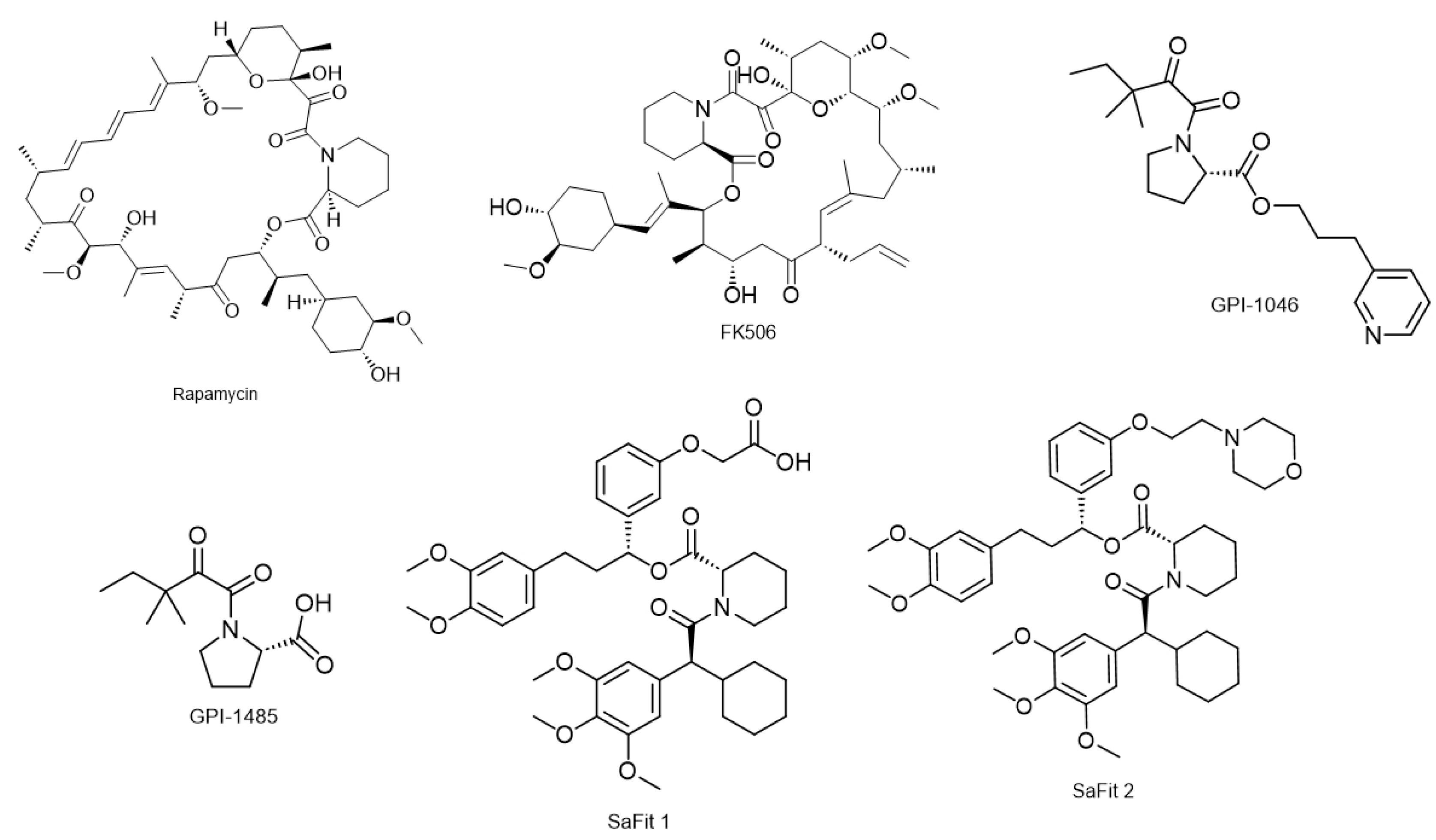
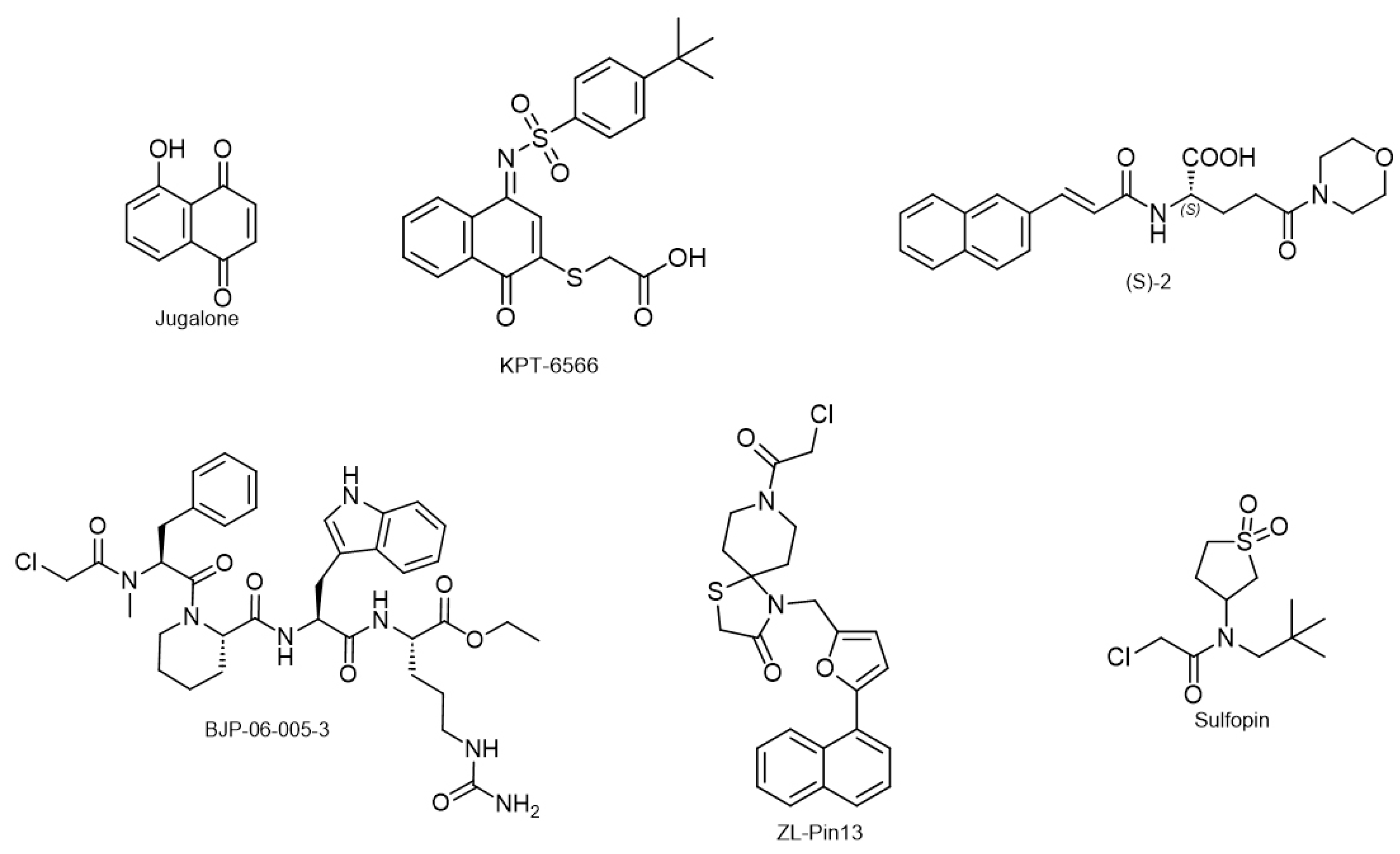
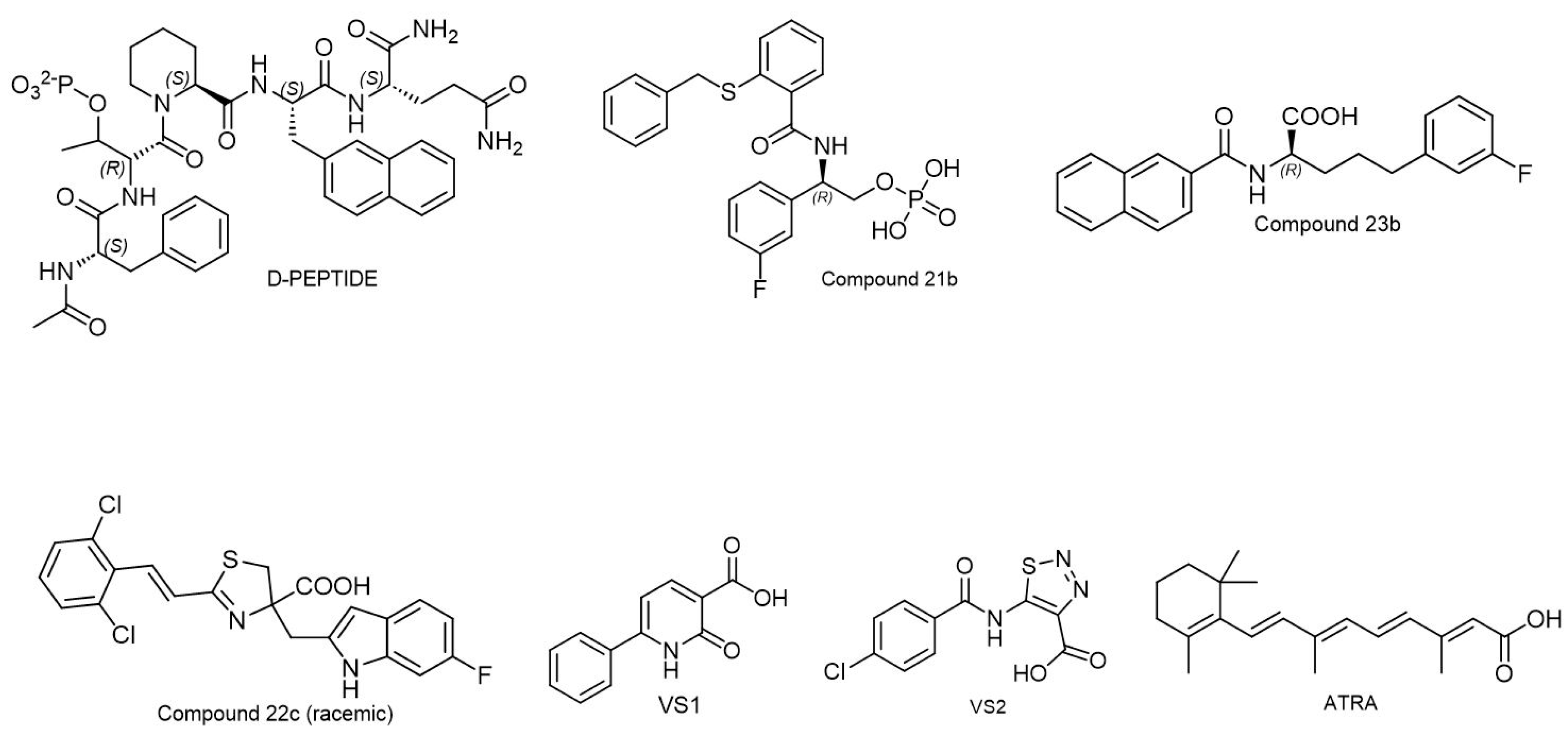

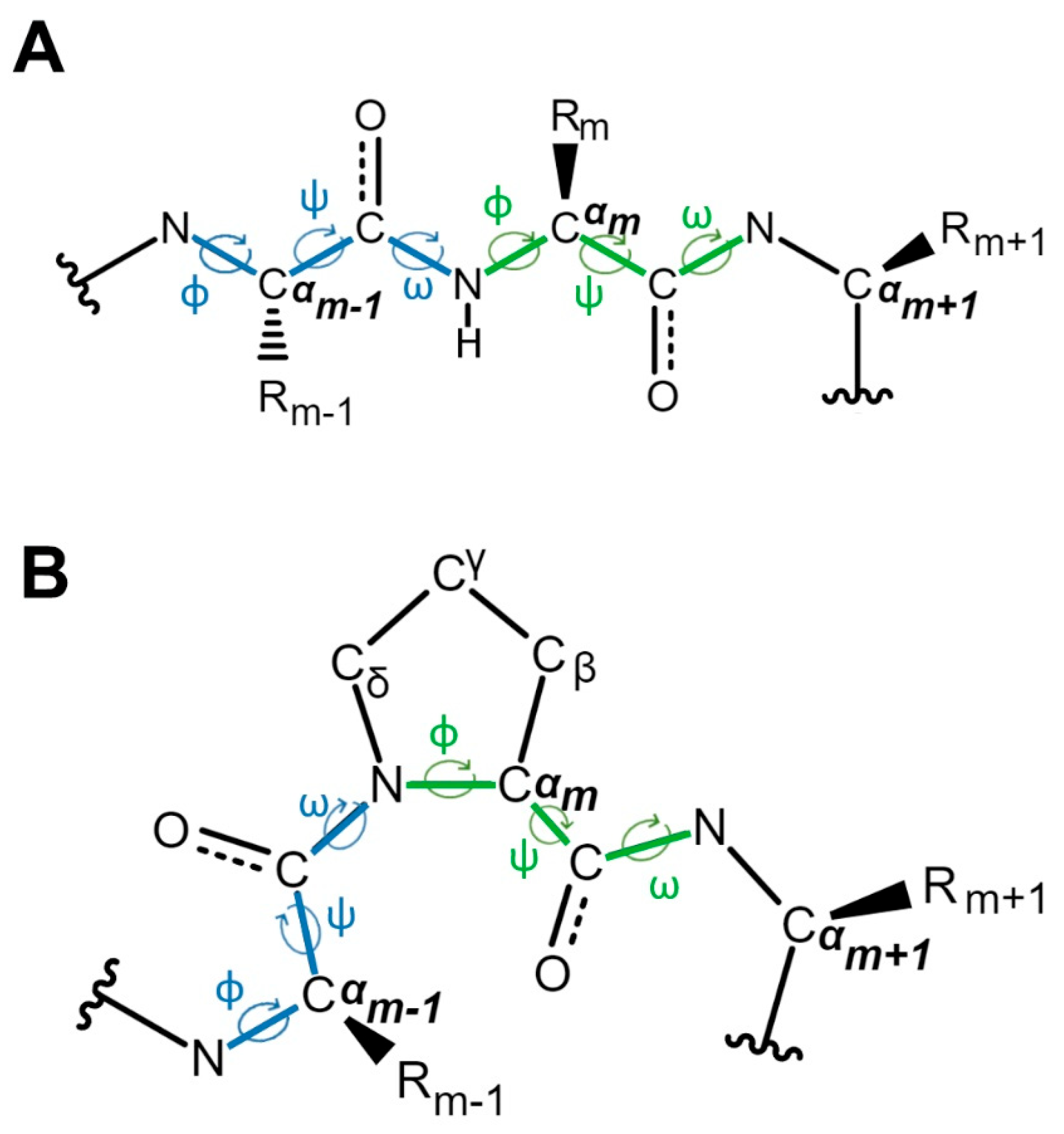{kind=link}
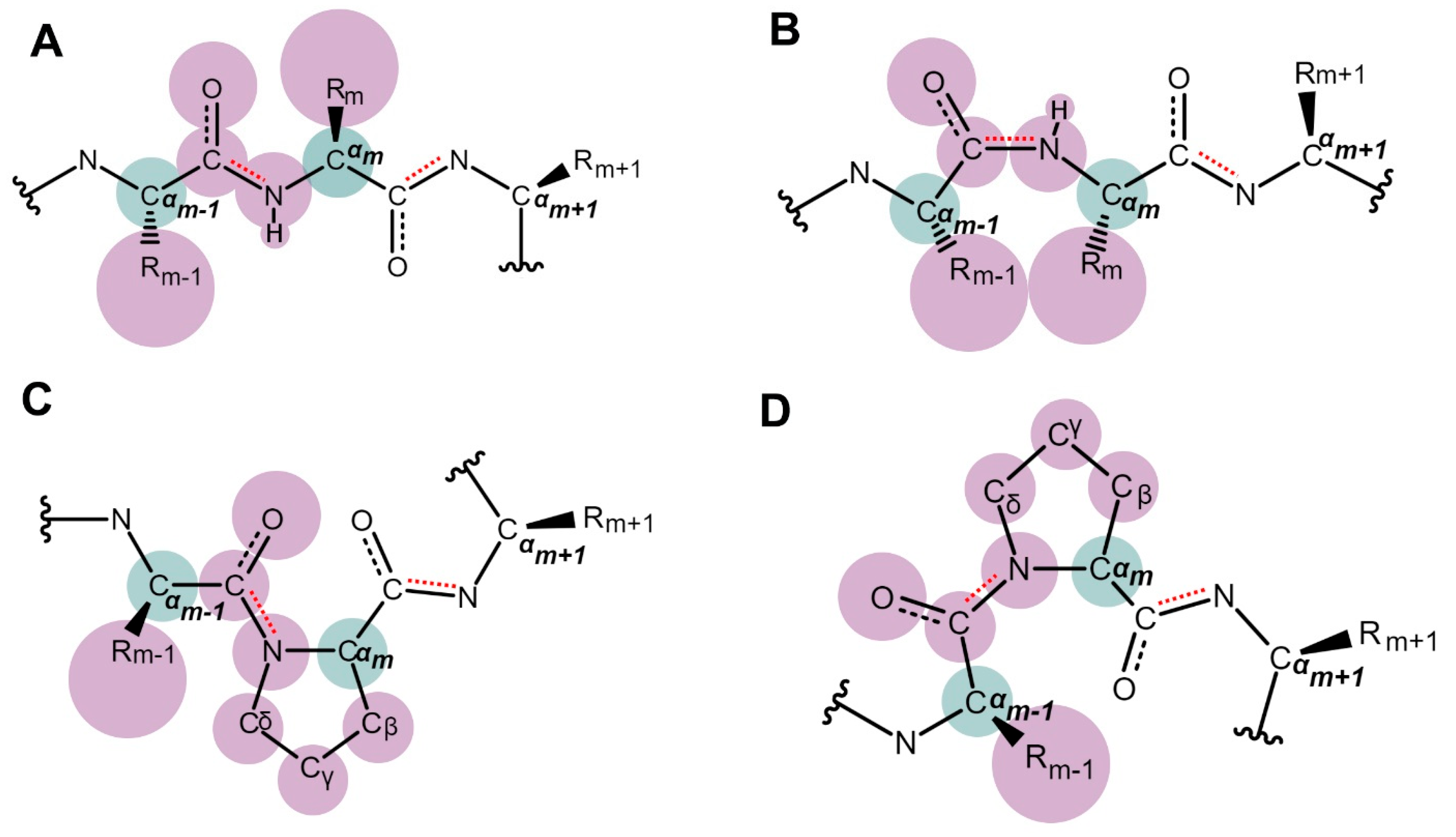{kind=link}
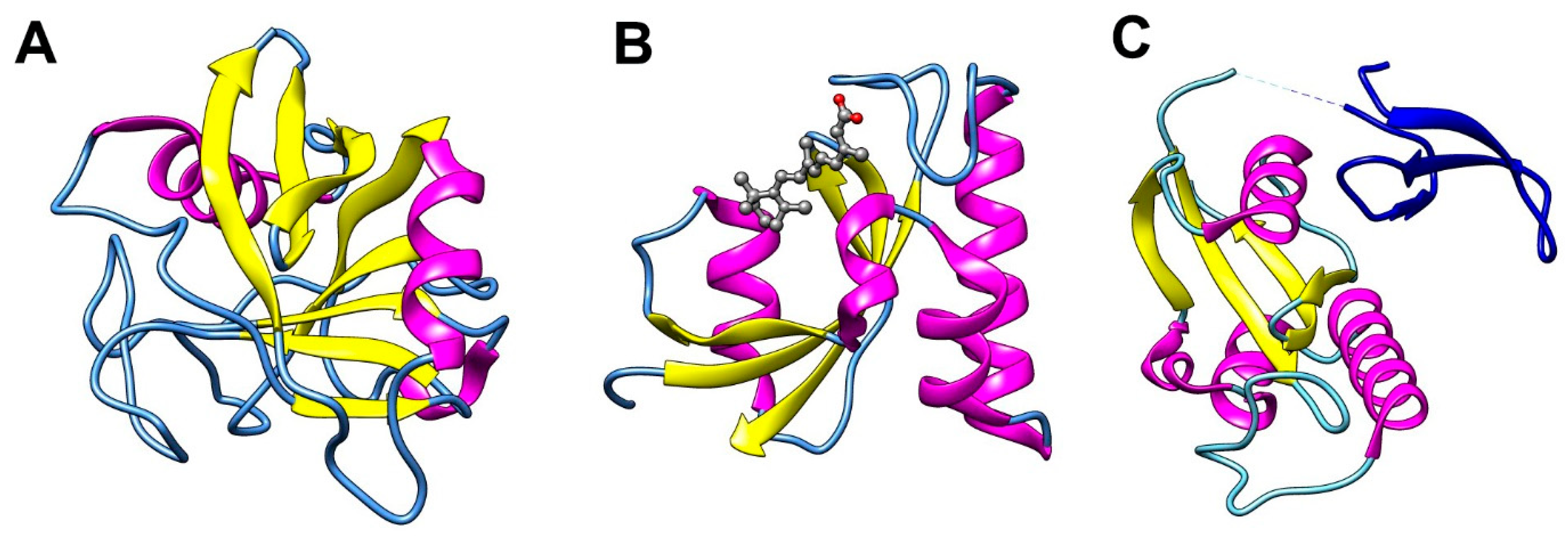{kind=link}
{kind=link}
{kind=link}
{kind=link}
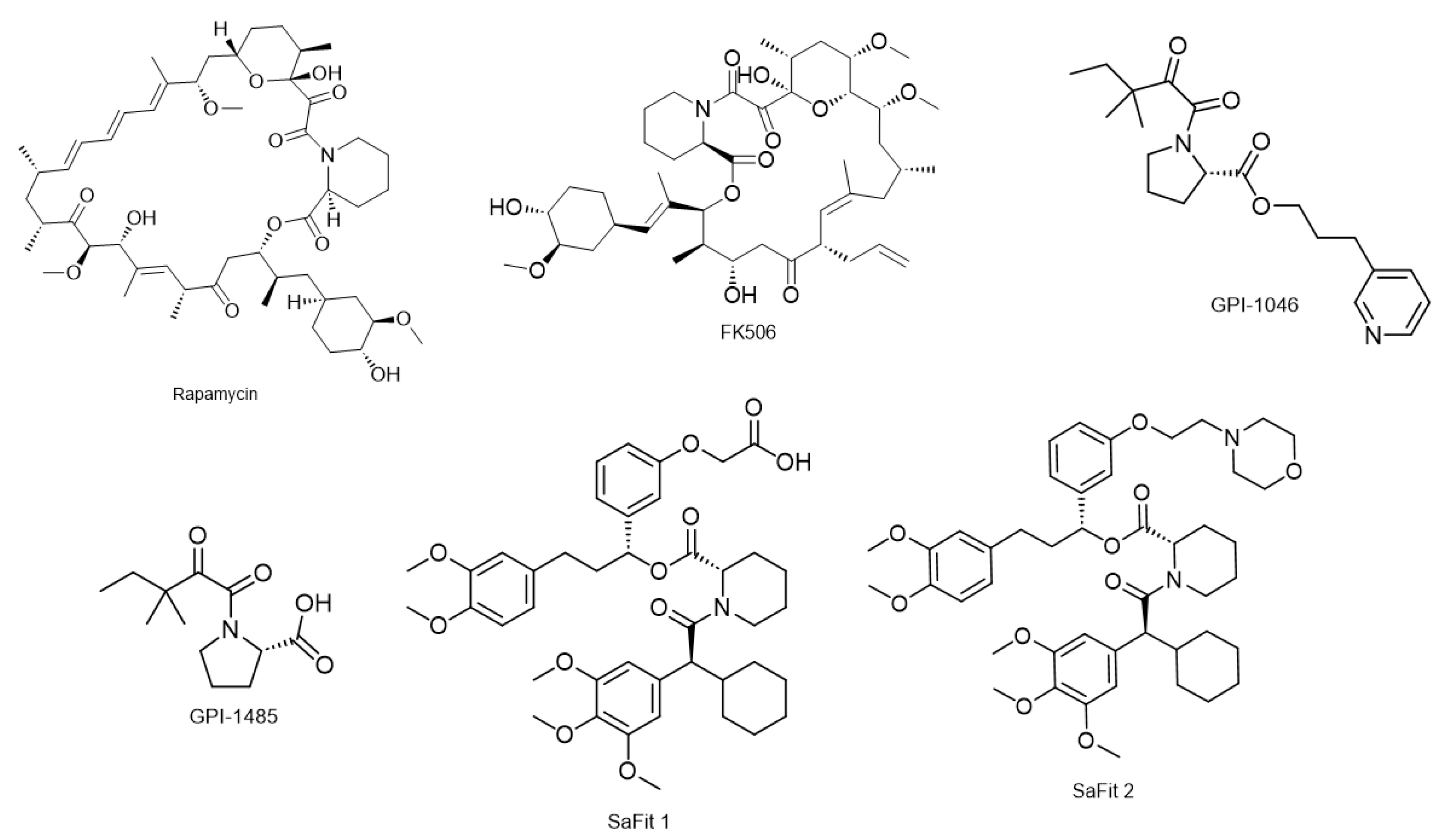{kind=link}
{kind=link}
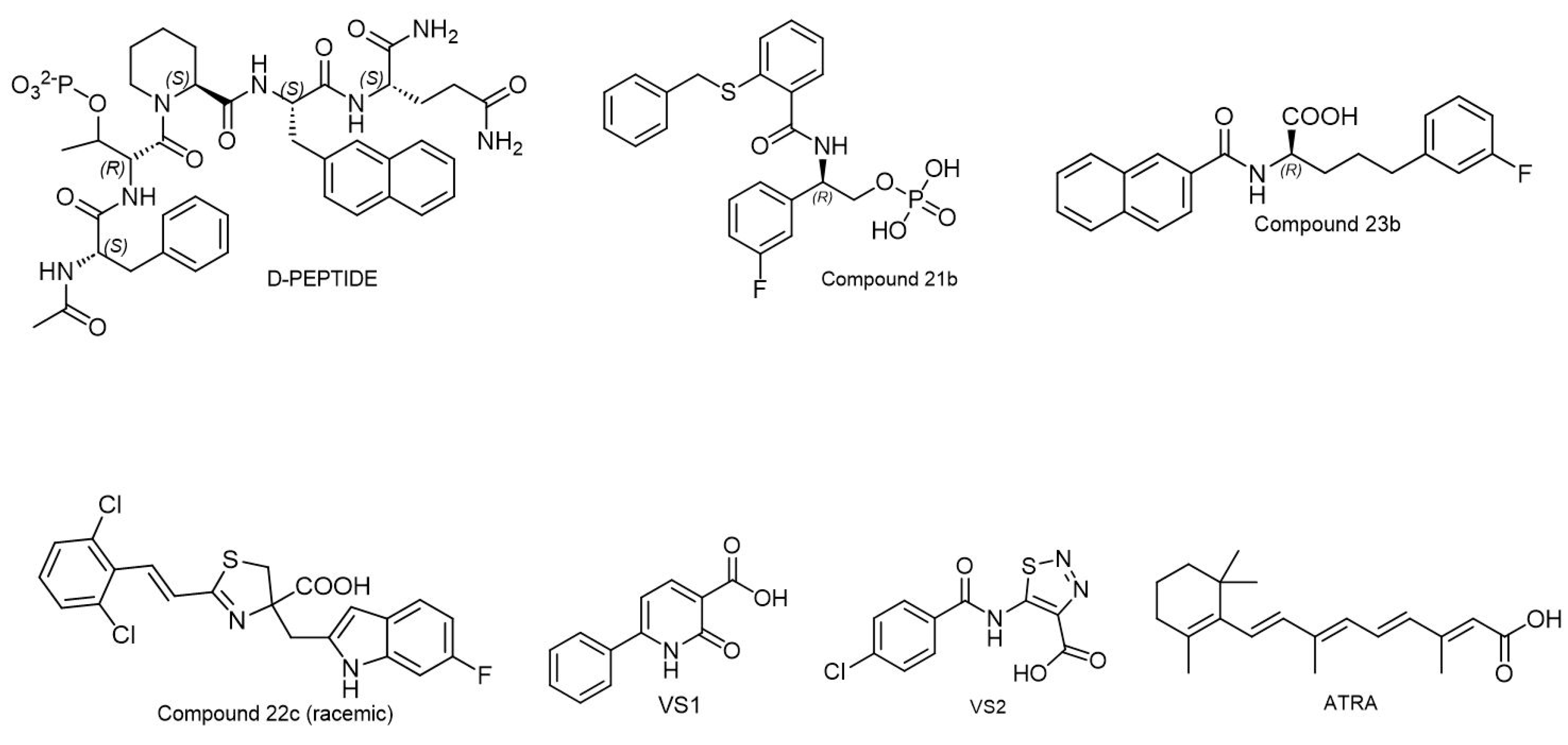{kind=link}
| Prolyl Isomerase | Protein Size | Preferred Proline Site | Triggers | Substrate Proteins | Subcellular Localization |
|---|---|---|---|---|---|
| FK506 binding proteins | 12–52 kDa | Xaa-Pro | Ligand binding, change in local environment of the protein such as pH | Tau APP, ryanodine receptor, Inositol 1,4,5-triphosphate receptor, TGF-β, heat-shock proteins, nuclear transport receptor including androgen receptor | Cytoplasm (primary), endoplasm reticulum (ER), and nucleus |
| Cyclophilins | 18–45 kDa | Xaa-Pro | Ligand binding, change in local environment of the protein such as pH | HIV1 capsid, NS5A of hepatitis C virus, heat -shock proteins (HSP90), α-synuclein | Cytosol, ER, mitochondria and nucleus |
| Parvulins | 14, 17, and 18 kDa | pSer/pThr-Pro (Pin1) Arg/Leu-Pro (Par14 and Par17) | Phosphorylation on Ser or Thr proceeding to Pro residue (Pin1) Arginine or leucine residues (Par14 and Par17) | ATR, p53, CDC25, α-synuclein, survivin, β-catenin, NF-kβ, Cyclin D1, C-Jun, RNA pol II | Nucleus (Pin1) Nucleus and cytosol (Par14) Mitochondria (Par17) |
| Inhibitors | Inhibitor Target | IC50 or Ki | Inhibitor Type | Reference |
|---|---|---|---|---|
| Cyclosporine | Cyclophilins | 420 ± 56 nM | Immunosuppressive | Sedrani et al., 2003 [226] |
| Debio-025 | 0.18 ± 0.03 nM | Non-Immunosuppressive Cyclosporine analog | Ptak et al., 2008 [227], Paeshuyse et al., 2006 [228] | |
| NIM811 | 0.66 μM | Non-Immunosuppressive Cyclosporine analog | Ma et al., 2006 [229] | |
| CRV431 | 2.5 nM (CypA) | Non-Immunosuppressive Cyclosporine analog | Kuo et al., 2019 [230] | |
| SCY-635 | 1.84 μM | Non-Immunosuppressive Cyclosporine analog | Hopkins et al., 2010 [231] | |
| Sanglifehrin A | 6.9 ± 0.9 nM | Immunosuppressive Sanglifehrin | Sedrani et al., 2003 [226] | |
| NV651 | 6.3 nM | Non-Immunosuppressive Sanglifehrin analog | Serrano et al., 2021 [232] | |
| NV556 | Non-Immunosuppressive Sanglifehrin analog | Kuo et al., 2019 [230], Serrano et al., 2019 [233] | ||
| Rapamycin | FKBPs | 0.1 nM | Immunosuppressive | Kolos et al., 2018 [61] |
| FK506 | 1 nM | Immunosuppressive | Kolos et al., 2018 [61] | |
| GPI-1485 | Immunosuppressive | Kolos et al., 2018 [61] | ||
| SaFit1 | 4 nM | Non-Immunosuppressive | Gaali et al., 2015 [234] | |
| SaFit2 | 8 nM | Non-Immunosuppressive | Gaali et al., 2015 [234] | |
| Juglone | Pin1 | 7.68 μM | Covalent inhibitor | Zhang et al., 2015 [235] |
| KPT-6566 | 0.64 μM | Covalent inhibitor | Campaner et al., 2017 [236] | |
| (S)-2 | 3.2 μM | Covalent inhibitor | Ieda et al., 2010 [237] | |
| BJP-06-005-3 | 48 nM | Non-covalent inhibitor | Pinch et al., 2020 [238] | |
| ZL-Pin13 | 0.067 ± 0.03 μM | Covalent inhibitor | Liu et al., 2022 [239] | |
| Sulfopin | 38 nM | Covalent inhibitor | Dubiella et al., 2021 [240] | |
| D-PEPTIDE | 20.4 ± 4.3 nM | Non-covalent inhibitor | Zhang et al., 2007 [55] | |
| Compound 21b | 6 nM | Non-covalent inhibitor | Guo et al., 2009 [241] | |
| Compound 23b | 890 nM | Non-covalent inhibitor | Dong et al., 2010 [242] | |
| Compound 22C | 196 nM | Non-covalent inhibitor | Guo et al., 2014 [243] | |
| VS1 | >100 μM | Non-covalent inhibitor | Poli et al., 2022 [244] | |
| VS2 | 19 ± 3 μM | Non-covalent inhibitor | Poli et al., 2022 [244] | |
| ATRA | 112 ± 10 μM | Non-covalent inhibitor | Poli et al., 2022 [244] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurung, D.; Danielson, J.A.; Tasnim, A.; Zhang, J.-T.; Zou, Y.; Liu, J.-Y. Proline Isomerization: From the Chemistry and Biology to Therapeutic Opportunities. Biology 2023, 12, 1008. https://doi.org/10.3390/biology12071008
Gurung D, Danielson JA, Tasnim A, Zhang J-T, Zou Y, Liu J-Y. Proline Isomerization: From the Chemistry and Biology to Therapeutic Opportunities. Biology. 2023; 12(7):1008. https://doi.org/10.3390/biology12071008
Chicago/Turabian StyleGurung, Deepti, Jacob A Danielson, Afsara Tasnim, Jian-Ting Zhang, Yue Zou, and Jing-Yuan Liu. 2023. "Proline Isomerization: From the Chemistry and Biology to Therapeutic Opportunities" Biology 12, no. 7: 1008. https://doi.org/10.3390/biology12071008
APA StyleGurung, D., Danielson, J. A., Tasnim, A., Zhang, J.-T., Zou, Y., & Liu, J.-Y. (2023). Proline Isomerization: From the Chemistry and Biology to Therapeutic Opportunities. Biology, 12(7), 1008. https://doi.org/10.3390/biology12071008