
Vaccination against Epstein–Barr Latent Membrane Protein 1 Protects against an Epstein–Barr Virus-Associated B Cell Model of Lymphoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Plasmid Construction and Cell Transfection
2.2. Western Blot and Immunofluorescence
2.3. 38C13 B Cell Transfection and Transformation
2.4. TMV-Peptide and Plasmid DNA Vaccine Preparation
2.5. Vaccination and Immune Response Measurement
2.6. Tumor Challenge
2.7. Statistical Analysis
3. Results
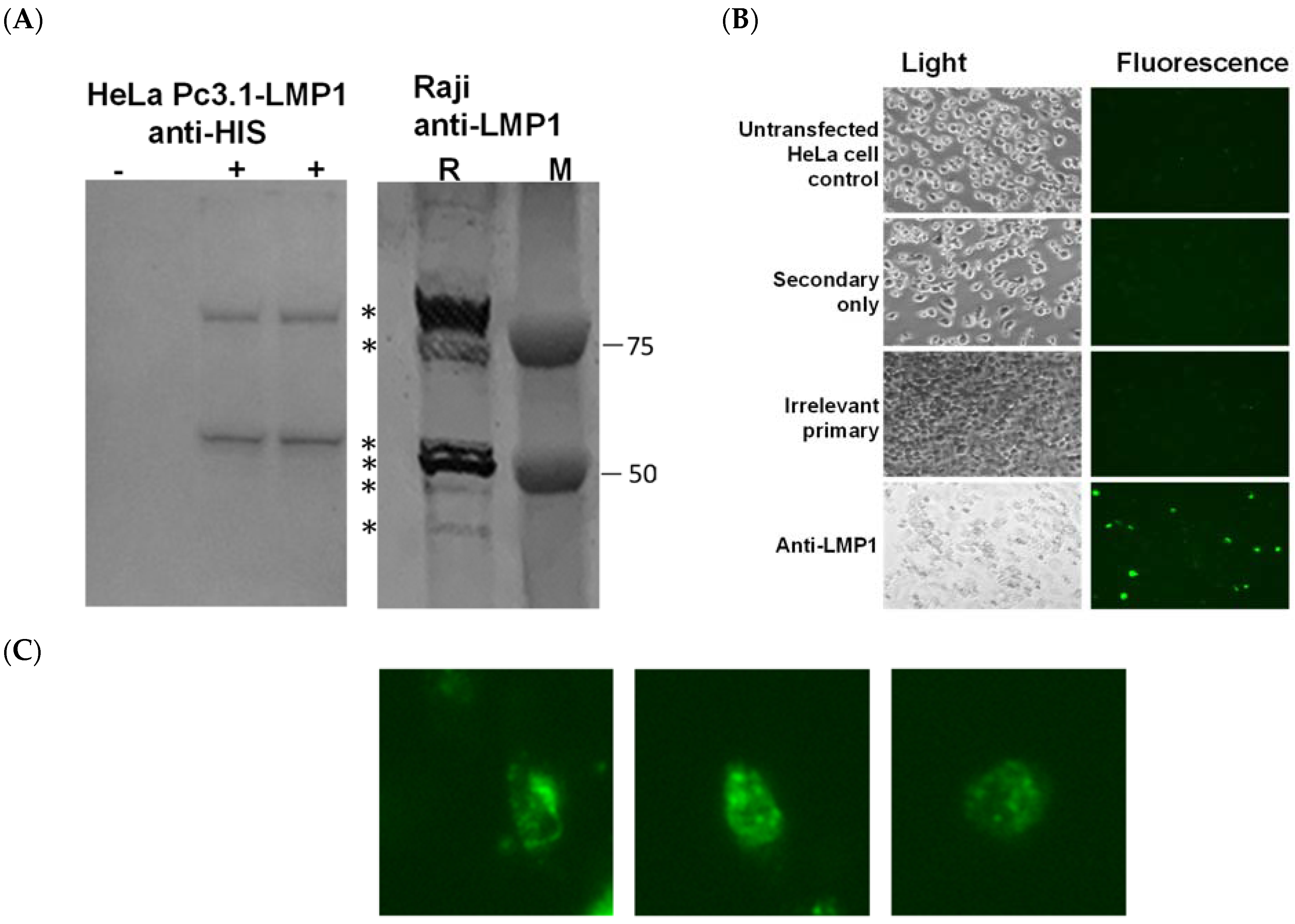
3.1. Validation of pc3.1-LMP1 Expression
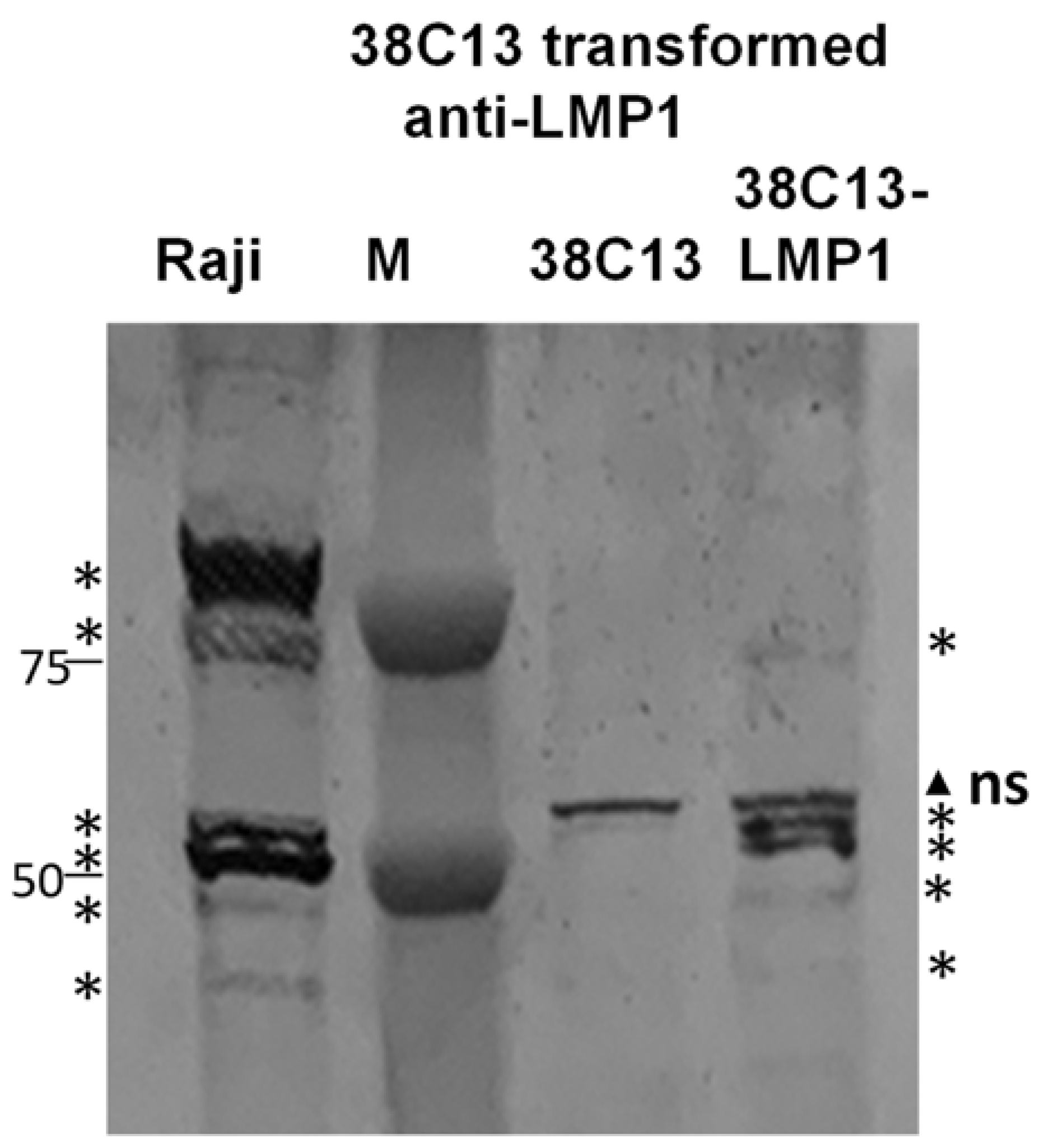
3.2. Transformation of 38C13 Mouse B Cells
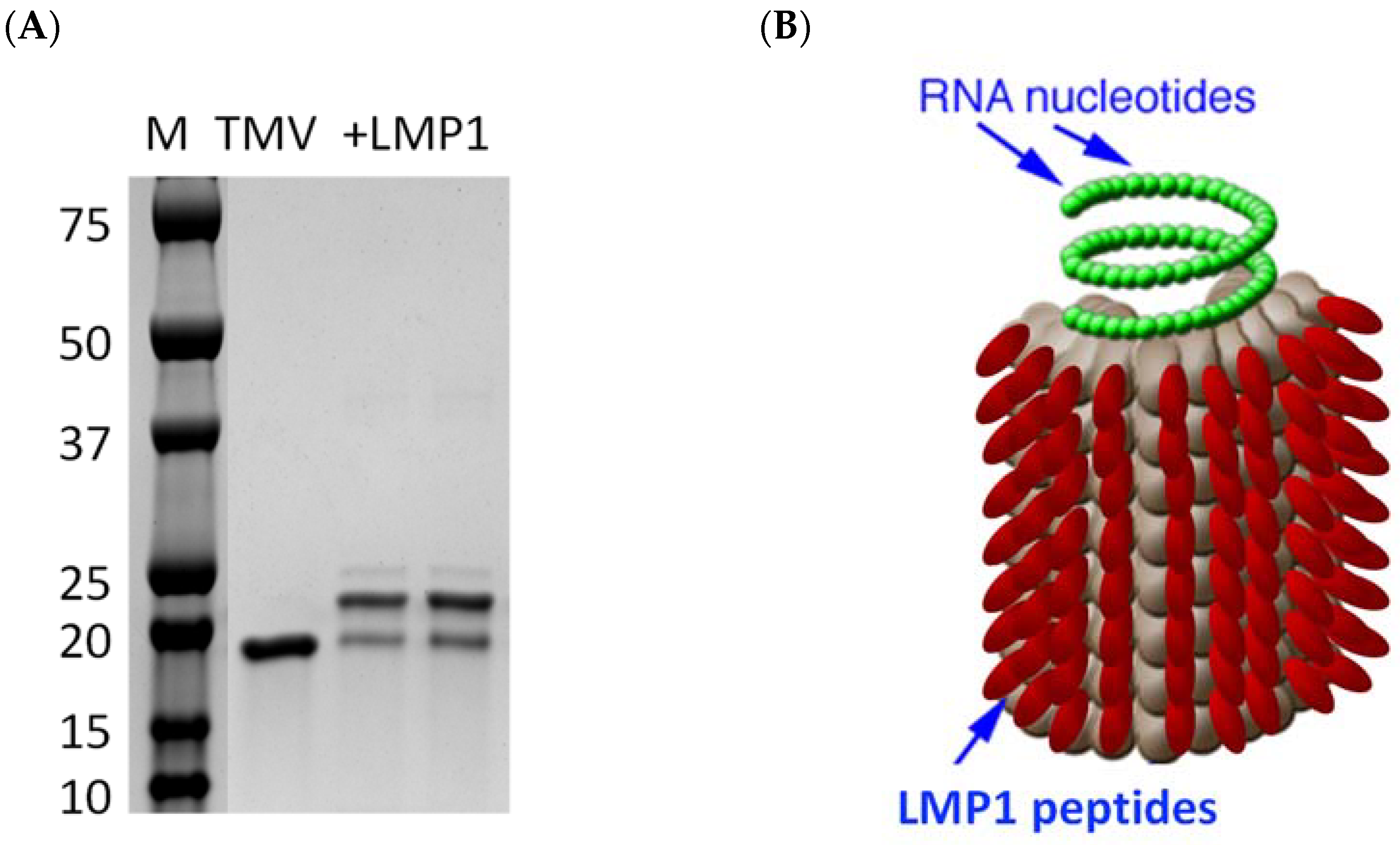
3.3. Creation of an LMP1 Peptide Vaccine
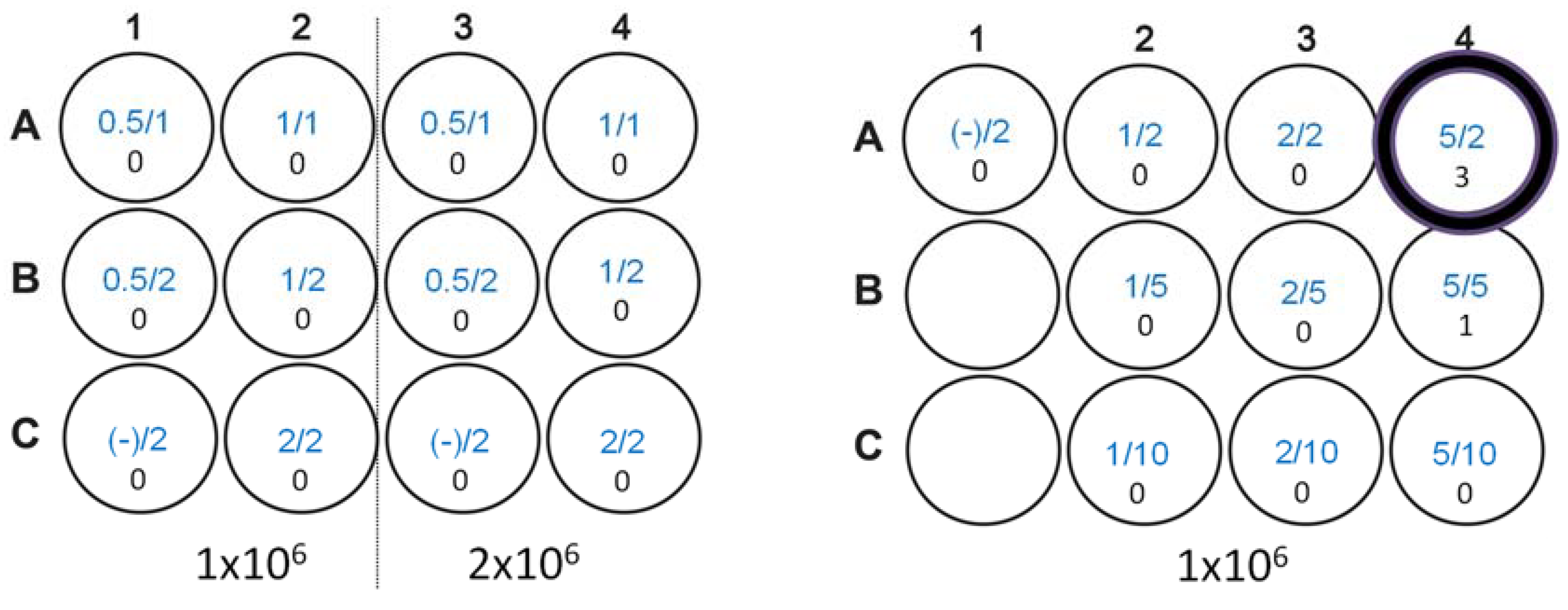
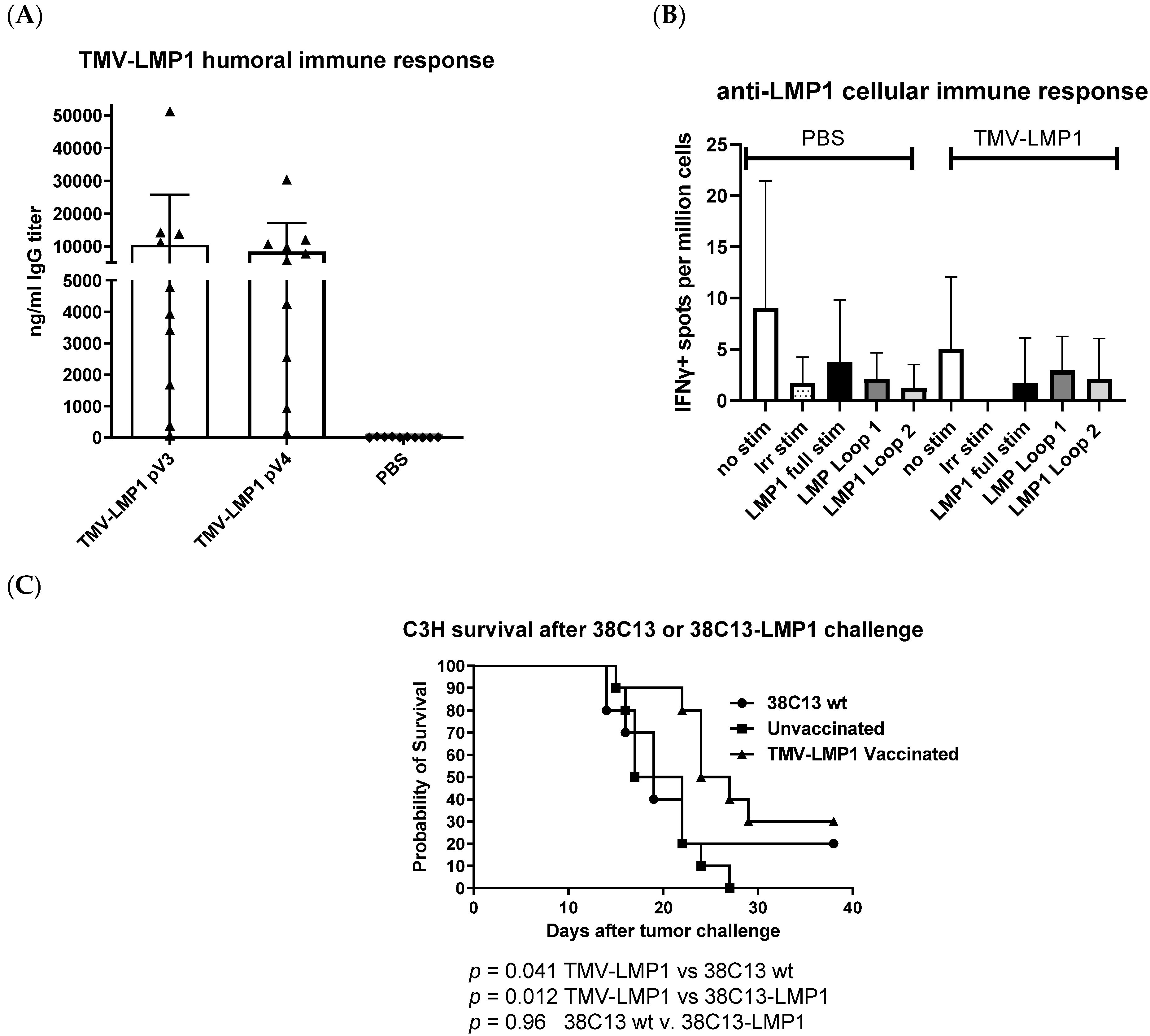
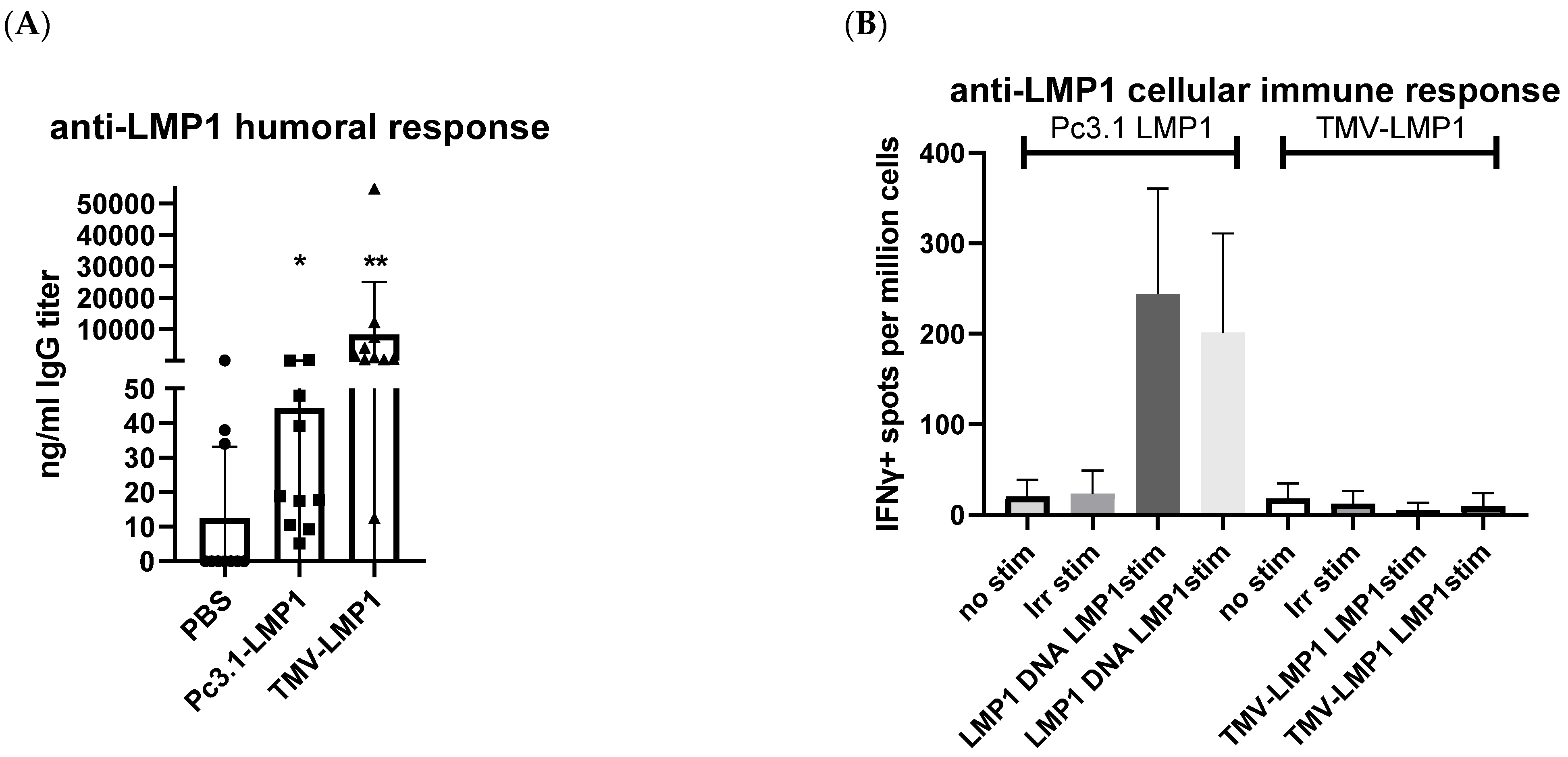
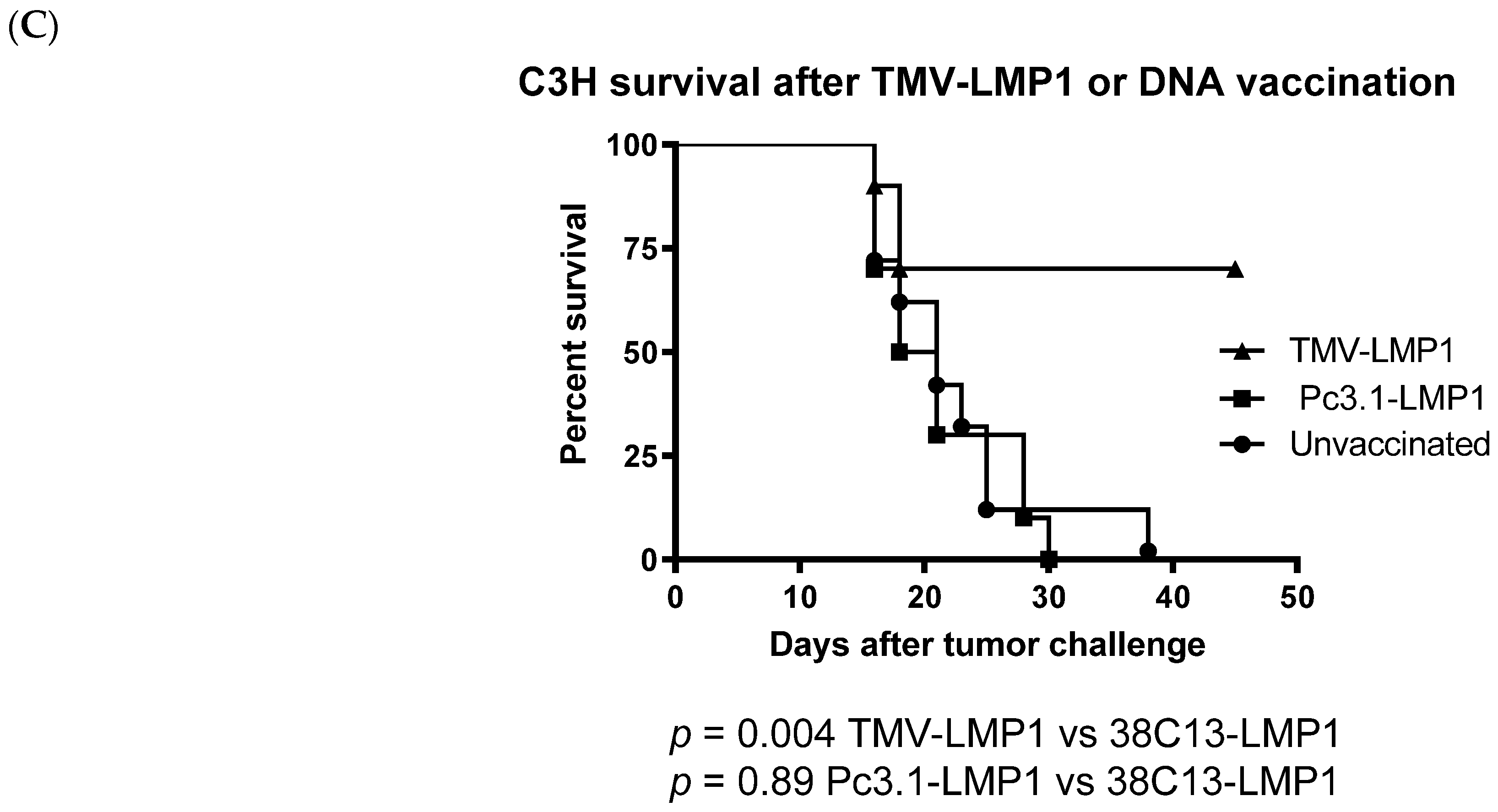
3.4. Immunization, Immune Analysis, and Tumor Challenge
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, E.; Akahane, M.; Kiryu, S.; Kato, N.; Yoshikawa, T.; Hayashi, N.; Aoki, S.; Minami, M.; Uozaki, H.; Fukayama, M.; et al. Spectrum of Epstein-Barr virus-related diseases: A pictorial review. Jpn. J. Radiol. 2009, 27, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Huang, Y.; Li, K.; Luo, R.; Cai, M.; Yun, J. Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein-Barr Virus-Associated Malignancies. Viruses 2022, 14, 1017. [Google Scholar] [CrossRef]
- Wong, Y.; Meehan, M.T.; Burrows, S.R.; Doolan, D.L.; Miles, J.J. Estimating the global burden of Epstein-Barr virus-related cancers. J. Cancer Res. Clin. Oncol. 2022, 148, 31–46. [Google Scholar] [CrossRef]
- Soldan, S.S.; Lieberman, P.M. Epstein-Barr virus and multiple sclerosis. Nat. Rev. Microbiol. 2023, 21, 51–64. [Google Scholar] [CrossRef]
- Saha, A.; Robertson, E.S. Epstein-Barr virus-associated B-cell lymphomas: Pathogenesis and clinical outcomes. Clin. Cancer Res. 2011, 17, 3056–3063. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein–Barr virus-associated lymphomas. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef] [Green Version]
- Williams, H. Epstein-Barr virus: The impact of scientific advances on clinical practice. Blood 2005, 107, 862–869. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hu, C.-F.; Hou, J.-H.; Shao, Q.; Yan, L.-X.; Zhu, X.-F.; Zeng, Y.-X.; Shao, J.-Y. Epstein-Barr virus encoded latent membrane protein 1 regulates mTOR signaling pathway genes which predict poor prognosis of nasopharyngeal carcinoma. J. Transl. Med. 2010, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Park, Y.; Kang, J.; Kim, K.; Ko, Y.; Ryoo, B.; Lee, S.; Lee, S.; Koo, H.; Kim, W. The effect of Epstein–Barr virus status on clinical outcome in Hodgkin’s lymphoma. Ann. Hematol. 2006, 85, 463–468. [Google Scholar] [CrossRef]
- Hariwiyanto, B.; Sastrowiyoto, S.; Mubarika, S.; Salugu, M. LMP1 and LMP2 may be prognostic factors for outcome of therapy in nasopharyngeal cancers in Indonesia. Asian Pac. J. Cancer Prev. 2010, 11, 763–766. [Google Scholar]
- Zhao, Y.; Wang, Y.; Zeng, S.; Hu, X. LMP1 expression is positively associated with metastasis of nasopharyngeal carcinoma: Evidence from a meta-analysis. J. Clin. Pathol. 2012, 65, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.P.; Arcipowski, K.M.; Bishop, G.A. Differential B-lymphocyte regulation by CD40 and its viral mimic, latent membrane protein 1. Immunol. Rev. 2010, 237, 226–248. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, M.; Izumi, K.M.; Kieff, E. Epstein–Barr virus latent-infection membrane proteins are palmitoylated and raft-associated: Protein 1 binds to the cytoskeleton through TNF receptor cytoplasmic factors. Proc. Natl. Acad. Sci. USA 2001, 98, 4675–4680. [Google Scholar] [CrossRef]
- D’Souza, B.N.; Edelstein, L.C.; Pegman, P.M.; Smith, S.M.; Loughran, S.T.; Clarke, A.; Mehl, A.; Rowe, M.; Gelinas, C.; Walls, D. Nuclear Factor B-Dependent Activation of the Antiapoptotic bfl-1 Gene by the Epstein-Barr Virus Latent Membrane Protein 1 and Activated CD40 Receptor. J. Virol. 2004, 78, 1800–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebowitz, D.; Mannick, J.; Takada, K.; Kieff, E. Phenotypes of Epstein-Barr virus LMP1 deletion mutants indicate transmembrane and amino-terminal cytoplasmic domains necessary for effects in B-lymphoma cells. J. Virol. 1992, 66, 4612–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [Green Version]
- Vrzalikova, K.; Ibrahim, M.; Nagy, E.; Vockerodt, M.; Perry, T.; Wei, W.; Woodman, C.; Murray, P. Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma. Cancers 2018, 10, 285. [Google Scholar] [CrossRef] [Green Version]
- Rowe, M. Epstein-Barr virus (EBV)-associated lymphoproliferative disease in the SCID mouse model: Implications for the pathogenesis of EBV-positive lymphomas in man. J. Exp. Med. 1991, 173, 147–158. [Google Scholar] [CrossRef]
- Yajima, M.; Imadome, K.I.; Nakagawa, A.; Watanabe, S.; Terashima, K.; Nakamura, H.; Ito, M.; Shimizu, N.; Honda, M.; Yamamoto, N.; et al. A New Humanized Mouse Model of Epstein-Barr Virus Infection That Reproduces Persistent Infection, Lymphoproliferative Disorder, and Cell-Mediated and Humoral Immune Responses. J. Infect. Dis. 2008, 198, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Yajima, M.; Imadome, K.I.; Nakagawa, A.; Watanabe, S.; Terashima, K.; Nakamura, H.; Ito, M.; Shimizu, N.; Yamamoto, N.; Fujiwara, S. T Cell–Mediated Control of Epstein-Barr Virus Infection in Humanized Mice. J. Infect. Dis. 2009, 200, 1611–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr Virus: An Important Vaccine Target for Cancer Prevention. Sci. Transl. Med. 2011, 3, 107fs107. [Google Scholar] [CrossRef] [Green Version]
- Delbende, C.; Verwaerde, C.; Mougel, A.; Tranchand Bunel, D. Induction of Therapeutic Antibodies by Vaccination against External Loops of Tumor-Associated Viral Latent Membrane Protein. J. Virol. 2009, 83, 11734–11745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longnecker, R.; Druker, B.; Roberts, T.M.; Kieff, E. An Epstein-Barr virus protein associated with cell growth transformation interacts with a tyrosine kinase. J. Virol. 1991, 65, 3681–3692. [Google Scholar] [CrossRef] [Green Version]
- Meij, P.; Leen, A.; Rickinson, A.B.; Verkoeijen, S.; Vervoort, M.B.H.J.; Bloemena, E.; Middeldorp, J.M. Identification and prevalence of CD8+ T-cell responses directed against Epstein-Barr virus-encoded latent membrane protein 1 and latent membrane protein 2. Int. J. Cancer 2002, 99, 93–99. [Google Scholar] [CrossRef]
- Lin, M.C.; Lin, Y.C.; Chen, S.T.; Young, T.H.; Lou, P.J. Therapeutic vaccine targeting Epstein-Barr virus latent protein, LMP1, suppresses LMP1-expressing tumor growth and metastasis in vivo. BMC Cancer 2017, 17, 18. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.A.; Corbo, T.A.; Wykoff-Clary, S.; Nguyen, L.V.; Smith, M.L.; Palmer, K.E.; Pogue, G.P. TMV-peptide fusion vaccines induce cell-mediated immune responses and tumor protection in two murine models. Vaccine 2006, 24, 6414–6423. [Google Scholar] [CrossRef]
- Kemnade, J.O.; Seethammagari, M.; Collinson-Pautz, M.; Kaur, H.; Spencer, D.M.; McCormick, A.A. Tobacco mosaic virus efficiently targets DC uptake, activation and antigen-specific T cell responses in vivo. Vaccine 2014, 32, 4228–4233. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.A.; Corbo, T.A.; Wykoff-Clary, S.; Palmer, K.E.; Pogue, G.P. Chemical conjugate TMV-peptide bivalent fusion vaccines improve cellular immunity and tumor protection. Bioconjugate Chem. 2006, 17, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Lindbo, J.A.; Dillard-Telm, S.; Brosio, P.M.; Lasnik, A.B.; McCormick, A.A.; Nguyen, L.V.; Palmer, K.E. Modified tobacco mosaic virus particles as scaffolds for display of protein antigens for vaccine applications. Virology 2006, 348, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattnaik, A.; Sahoo, B.R.; Struble, L.R.; Borgstahl, G.E.O.; Zhou, Y.; Franco, R.; Barletta, R.G.; Osorio, F.A.; Petro, T.M.; Pattnaik, A.K. A Ferritin Nanoparticle-Based Zika Virus Vaccine Candidate Induces Robust Humoral and Cellular Immune Responses and Protects Mice from Lethal Virus Challenge. Vaccines 2023, 11, 821. [Google Scholar] [CrossRef] [PubMed]
- McCormick, A.A.; Kumagai, M.H.; Hanley, K.; Turpen, T.H.; Hakim, I.; Grill, L.K.; Tuse, D.; Levy, S.; Levy, R. Rapid production of specific vaccines for lymphoma by expression of the tumor-derived single-chain Fv epitopes in tobacco plants. Proc. Natl. Acad. Sci. USA 1999, 96, 703–708. [Google Scholar] [CrossRef]
- Ling Yong, C.L.; Siak-Wei Ow, D.; Tandiono, T.; Mei Heng, L.L.; Kwok-Keung Chan, K.; Ohl, C.D.; Klaseboer, E.; Ohl, S.W.; Boon-Hwa Choo, A. Microbubble-mediated sonoporation for highly efficient transfection of recalcitrant human B- cell lines. Biotechnol. J. 2014, 9, 1081–1087. [Google Scholar] [CrossRef]
- Malik, A.; Houghten, R.; Corradin, G.; Buus, S.; Berzofsky, J.A.; Hoffman, S.L. Identification of a nonameric H-2Kk-restricted CD8+ cytotoxic T lymphocyte epitope on the Plasmodium falciparum circumsporozoite protein. Infect. Immun. 1995, 63, 1955–1959. [Google Scholar] [CrossRef] [Green Version]
- Millrain, M.; Scott, D.; Addey, C.; Dewchand, H.; Ellis, P.; Ehrmann, I.; Mitchell, M.; Burgoyne, P.; Simpson, E.; Dyson, J. Identification of the immunodominant HY H2-D(k) epitope and evaluation of the role of direct and indirect antigen presentation in HY responses. J. Immunol. 2005, 175, 7209–7217. [Google Scholar] [CrossRef] [Green Version]
- Vitiello, A.; Sette, A.; Yuan, L.; Farness, P.; Southwood, S.; Sidney, J.; Chesnut, R.W.; Grey, H.M.; Livingston, B. Comparison of cytotoxic T lymphocyte responses induced by peptide or DNA immunization: Implications on immunogenicity and immunodominance. Eur. J. Immunol. 1997, 27, 671–678. [Google Scholar] [CrossRef]
- Le Page, C.; Genin, P.; Baines, M.G.; Hiscott, J. Interferon activation and innate immunity. Rev. Immunogenet. 2000, 2, 374–386. [Google Scholar]
- Lee, S.-H.; Danishmalik, S.N.; Sin, J.-I. DNA vaccines, electroporation and their applications in cancer treatment. Hum. Vaccines Immunother. 2015, 11, 1889–1900. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Lewis, A.E.; Robertson, E.S. Current Progress in EBV-Associated B-Cell Lymphomas. Adv. Exp. Med. Biol. 2017, 1018, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Hao, Y.; Wang, Z.; Zeng, Y. Constructing TC-1-GLUC-LMP2 Model Tumor Cells to Evaluate the Anti-Tumor Effects of LMP2-Related Vaccines. Viruses 2018, 10, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtak, K.; Perales-Puchalt, A.; Weiner, D.B. Novel Synthetic DNA Immunogens Targeting Latent Expressed Antigens of Epstein-Barr Virus Elicit Potent Cellular Responses and Inhibit Tumor Growth. Vaccines 2019, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, A.A.; Hussain, Z.F.; Hashmi, K.A.; Zafar, M.I.; Edhi, M.M.; Faridi, N.; Khan, M. Latent membrane protein 1 (LMP1) expression in Hodgkin lymphoma and its correlation with clinical and histologic parameters. World J. Surg. Oncol. 2017, 15, 89. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, X.; Cheng, C.; Jiang, S.; Ma, T.; Xu, L. Absence of MHC class Ⅱ molecules promotes natural killer cells activation in mice. Int. Immunopharmacol. 2020, 88, 106888. [Google Scholar] [CrossRef]
- Jiang, X.N.; Yu, B.H.; Yan, W.H.; Lee, J.; Zhou, X.Y.; Li, X.Q. Epstein-Barr virus-positive diffuse large B-cell lymphoma features disrupted antigen capture/presentation and hijacked T-cell suppression. Oncoimmunology 2020, 9, 1683346. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.J.; Carroll, W.; Kon, S.; Thielemans, K.; Rothbard, J.B.; Levy, S.; Levy, R. Idiotype vaccination against murine B cell lymphoma. Humoral and cellular responses elicited by tumor-derived immunoglobulin M and its molecular subunits. J. Immunol. 1987, 139, 2825–2833. [Google Scholar] [CrossRef]
- Kwak, L.W.; Young, H.A.; Pennington, R.W.; Weeks, S.D. Vaccination with syngeneic, lymphoma-derived immunoglobulin idiotype combined with granulocyte/macrophage colony-stimulating factor primes mice for a protective T-cell response. Proc. Natl. Acad. Sci. USA 1996, 93, 10972–10977. [Google Scholar] [CrossRef]
- Hsu, F.J.; Caspar, C.B.; Czerwinski, D.; Kwak, L.W.; Liles, T.M.; Syrengelas, A.; Taidi-Laskowski, B.; Levy, R. Tumor-specific idiotype vaccines in the treatment of patients with B- cell lymphoma--long-term results of a clinical trial. Blood 1997, 89, 3129–3135. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.A.; Reddy, S.; Reinl, S.J.; Cameron, T.I.; Czerwinski, D.; Vojdani, F.; Hanley, K.M.; Garger, S.J.; White, E.L.; Novak, J.; et al. Plant-produced Idiotype Vaccines for the Treatment of Non-Hodgkin’s Lymphoma: Safety and Immunogenicity in a Phase I Clinical Study. Proc. Natl. Acad. Sci. USA 2008, 105, 10131–10136. [Google Scholar] [CrossRef] [PubMed]
- Mallajosyula, J.K.; Jeevan, T.; Chickwamba, R.; Webby, R.J.; McCormick, A.A. A single dose TMV-HA vaccine protects mice from H5N1 influenza challenge. Int. J. Vaccine Res. 2016, 1, 6–11. [Google Scholar]
- Mallajosyula, J.K.; Hiatt, E.; Hume, S.; Johnson, A.; Jeevan, T.; Chikwamba, R.; Pogue, G.P.; Bratcher, B.; Haydon, H.; Webby, R.J.; et al. Single-dose monomeric HA subunit vaccine generates full protection from influenza challenge. Hum. Vaccines Immunother. 2013, 10, 586–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royal, J.M.; Simpson, C.A.; McCormick, A.A.; Phillips, A.; Hume, S.; Morton, J.; Shepherd, J.; Oh, Y.; Swope, K.; DeBeauchamp, J.L.; et al. Development of a SARS-CoV-2 Vaccine Candidate Using Plant-Based Manufacturing and a Tobacco Mosaic Virus-like Nano-Particle. Vaccines 2021, 9, 1347. [Google Scholar] [CrossRef]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Ruhl, J.; Leung, C.S.; Munz, C. Vaccination against the Epstein-Barr virus. Cell Mol. Life Sci. 2020, 77, 4315–4324. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Wang, W.; Erbe, A.K.; Hank, J.A.; Morris, Z.S.; Sondel, P.M. NK Cell-Mediated Antibody-Dependent Cellular Cytotoxicity in Cancer Immunotherapy. Front. Immunol. 2015, 6, 368. [Google Scholar] [CrossRef] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soo Hoo, W.I.; Higa, K.; McCormick, A.A. Vaccination against Epstein–Barr Latent Membrane Protein 1 Protects against an Epstein–Barr Virus-Associated B Cell Model of Lymphoma. Biology 2023, 12, 983. https://doi.org/10.3390/biology12070983
Soo Hoo WI, Higa K, McCormick AA. Vaccination against Epstein–Barr Latent Membrane Protein 1 Protects against an Epstein–Barr Virus-Associated B Cell Model of Lymphoma. Biology. 2023; 12(7):983. https://doi.org/10.3390/biology12070983
Chicago/Turabian StyleSoo Hoo, Wesley I., Kaylie Higa, and Alison A. McCormick. 2023. "Vaccination against Epstein–Barr Latent Membrane Protein 1 Protects against an Epstein–Barr Virus-Associated B Cell Model of Lymphoma" Biology 12, no. 7: 983. https://doi.org/10.3390/biology12070983
APA StyleSoo Hoo, W. I., Higa, K., & McCormick, A. A. (2023). Vaccination against Epstein–Barr Latent Membrane Protein 1 Protects against an Epstein–Barr Virus-Associated B Cell Model of Lymphoma. Biology, 12(7), 983. https://doi.org/10.3390/biology12070983