
Iron Metabolism in Cancer and Senescence: A Cellular Perspective


Abstract
:Simple Summary
Abstract
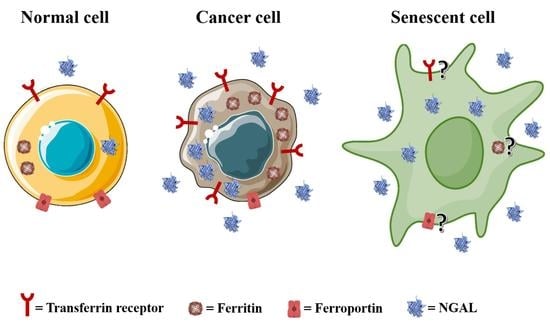
{kind=link}
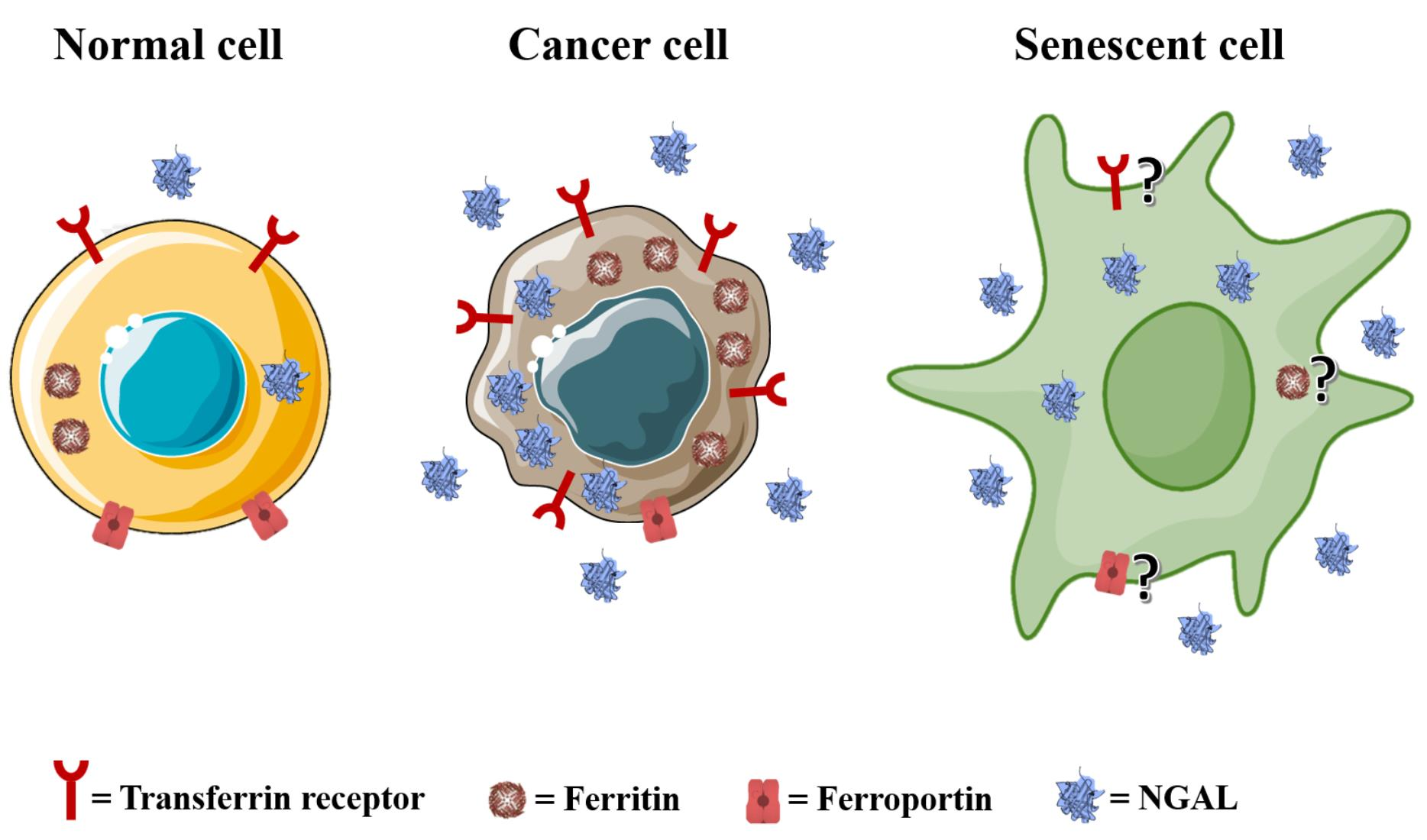
{kind=link}
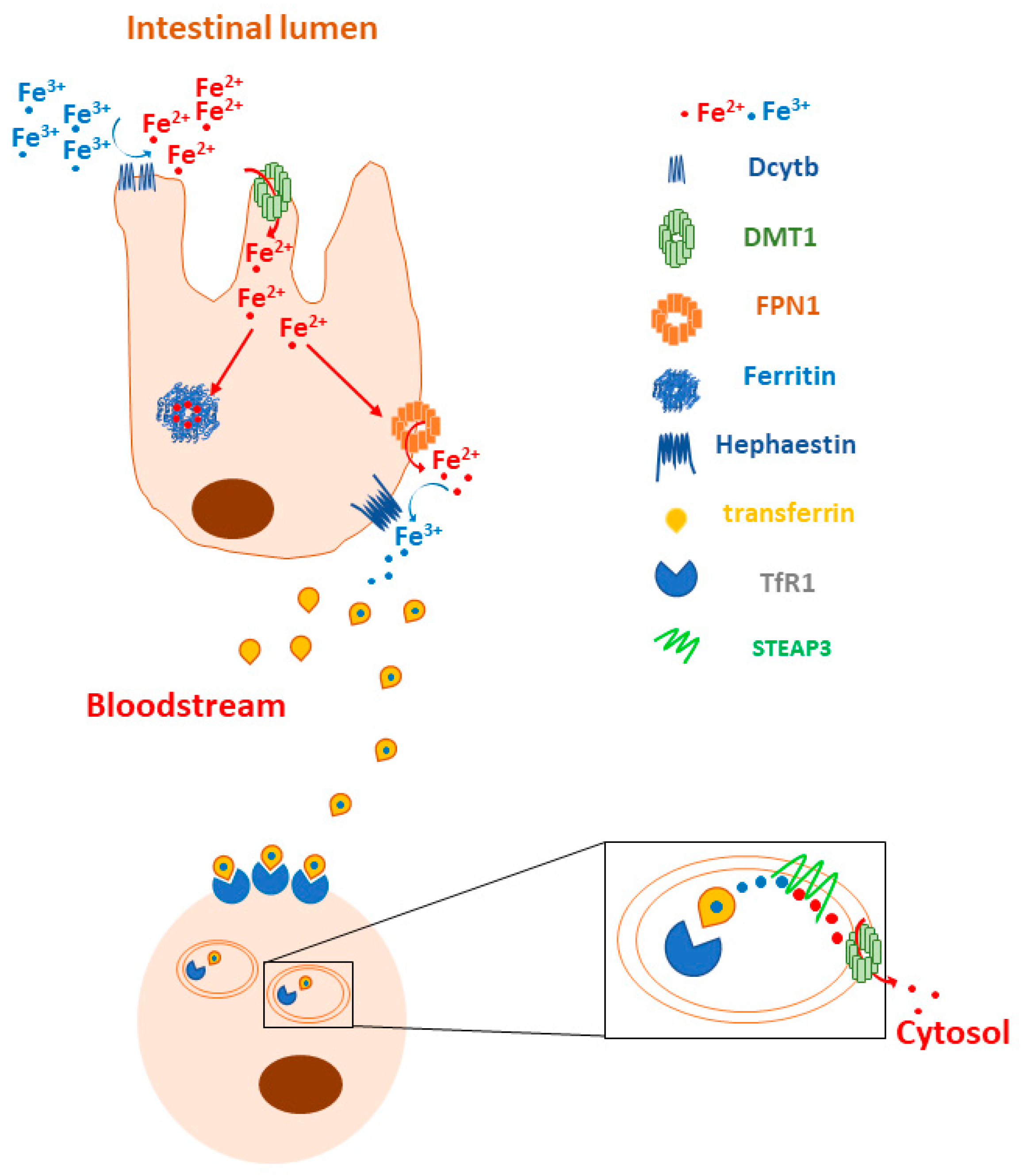
{kind=link}
{kind=link}
{kind=link}
1. Introduction
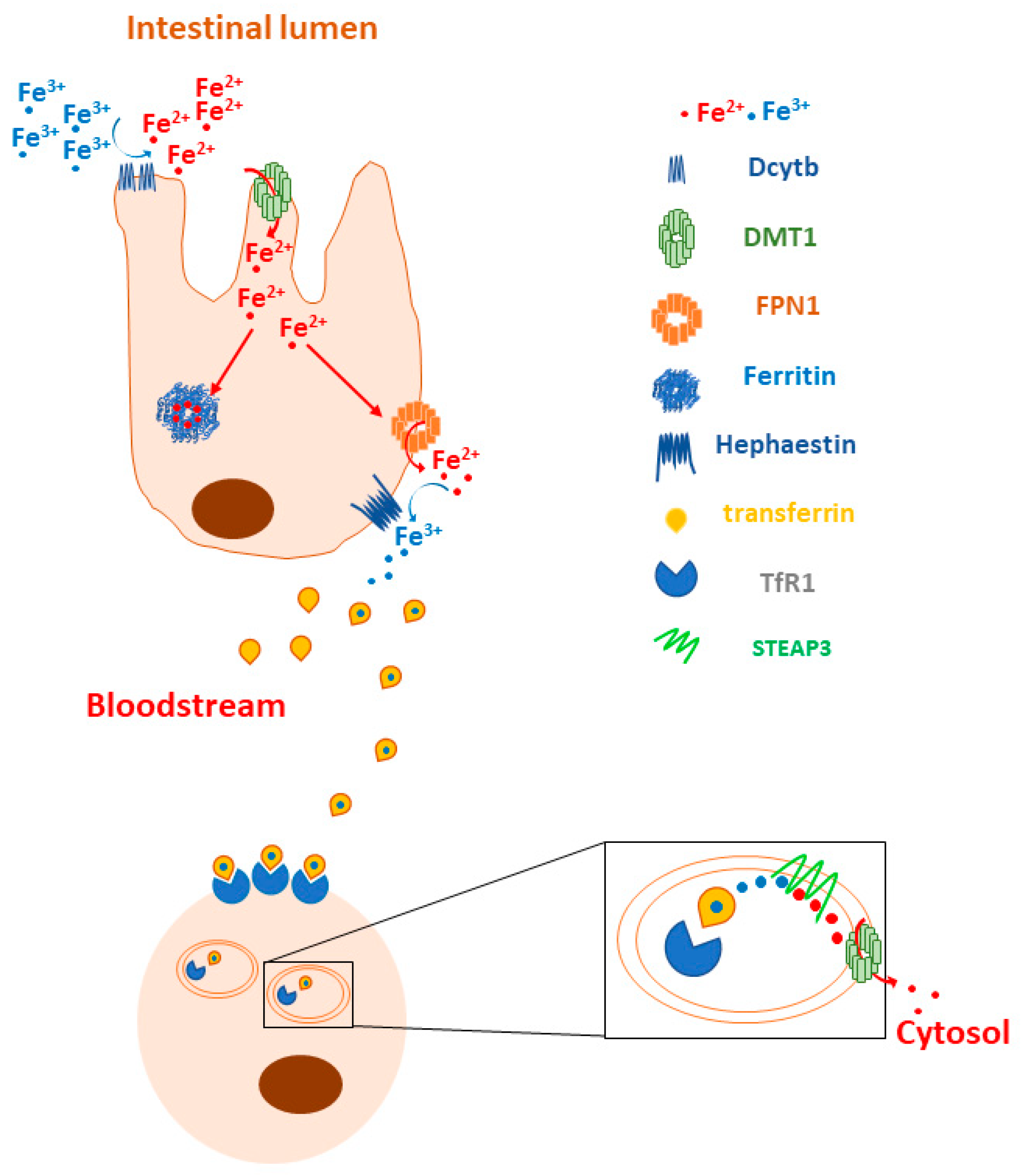
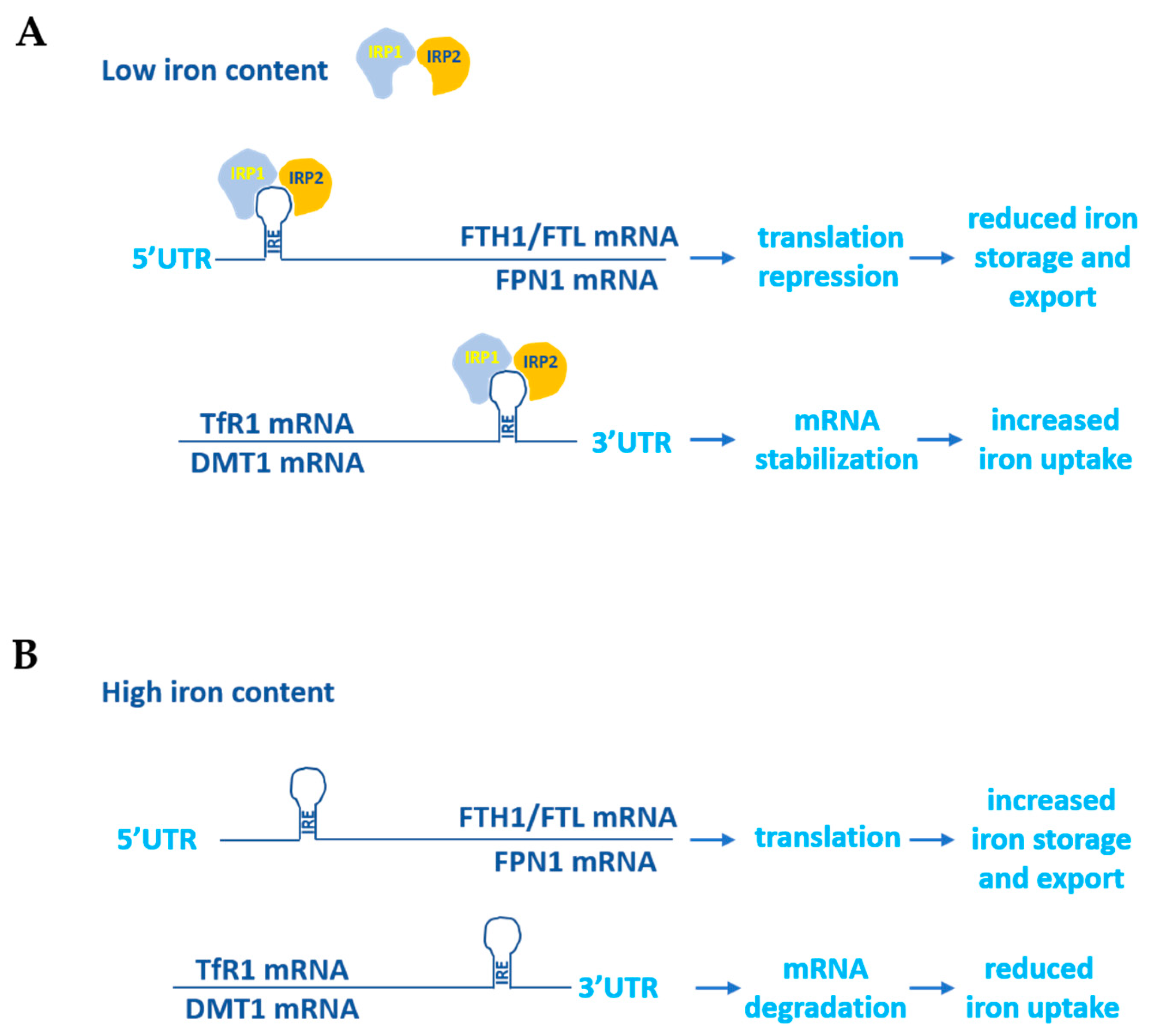
1.1. Iron Metabolism in Normal Cells
1.2. Iron in Cancer and Senescence
2. Key Iron Regulatory Proteins in Cancer and Senescence
2.1. Transferrin Receptor 1 and 2 (TfR1, TfR2)
2.2. Ferritin
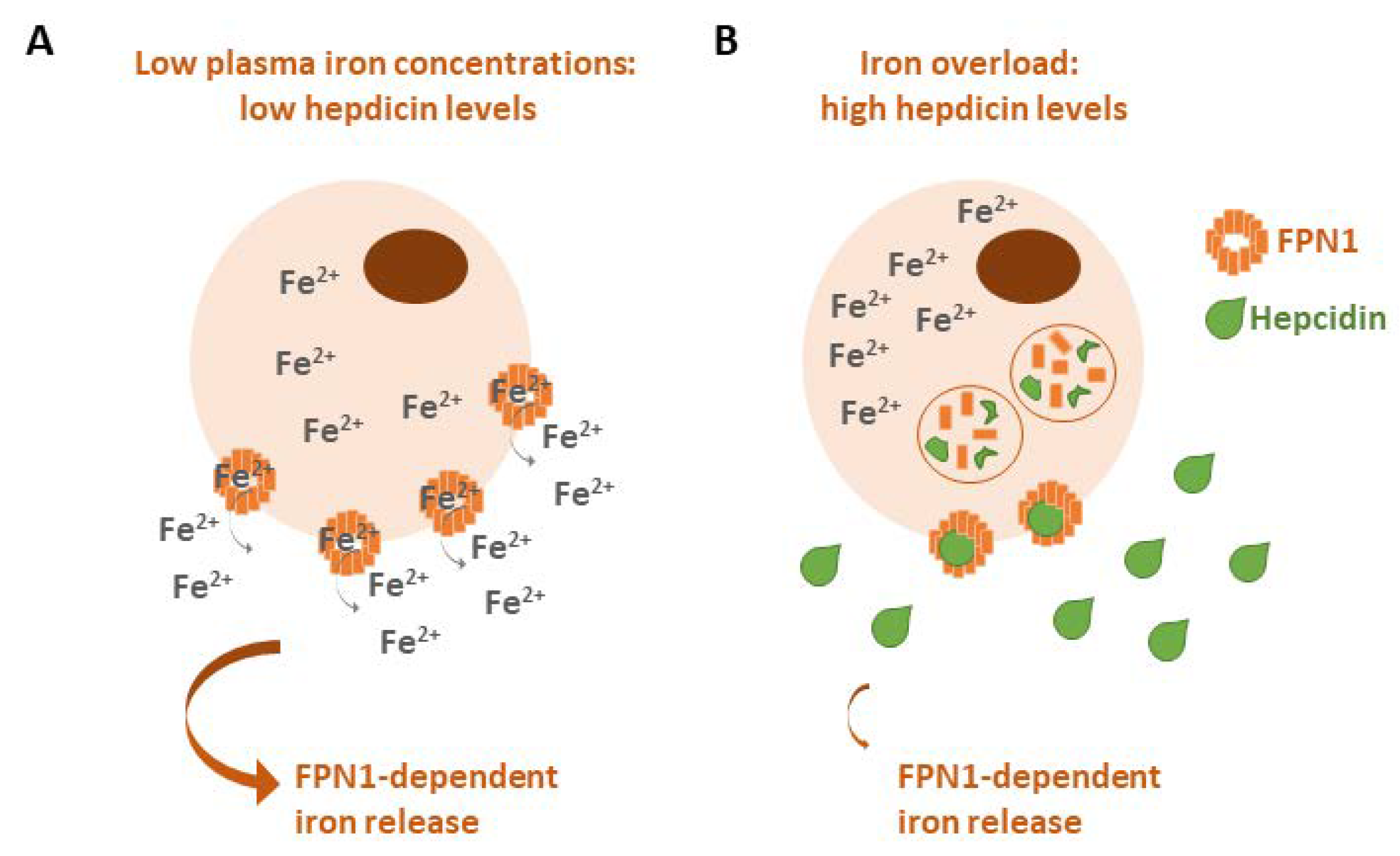
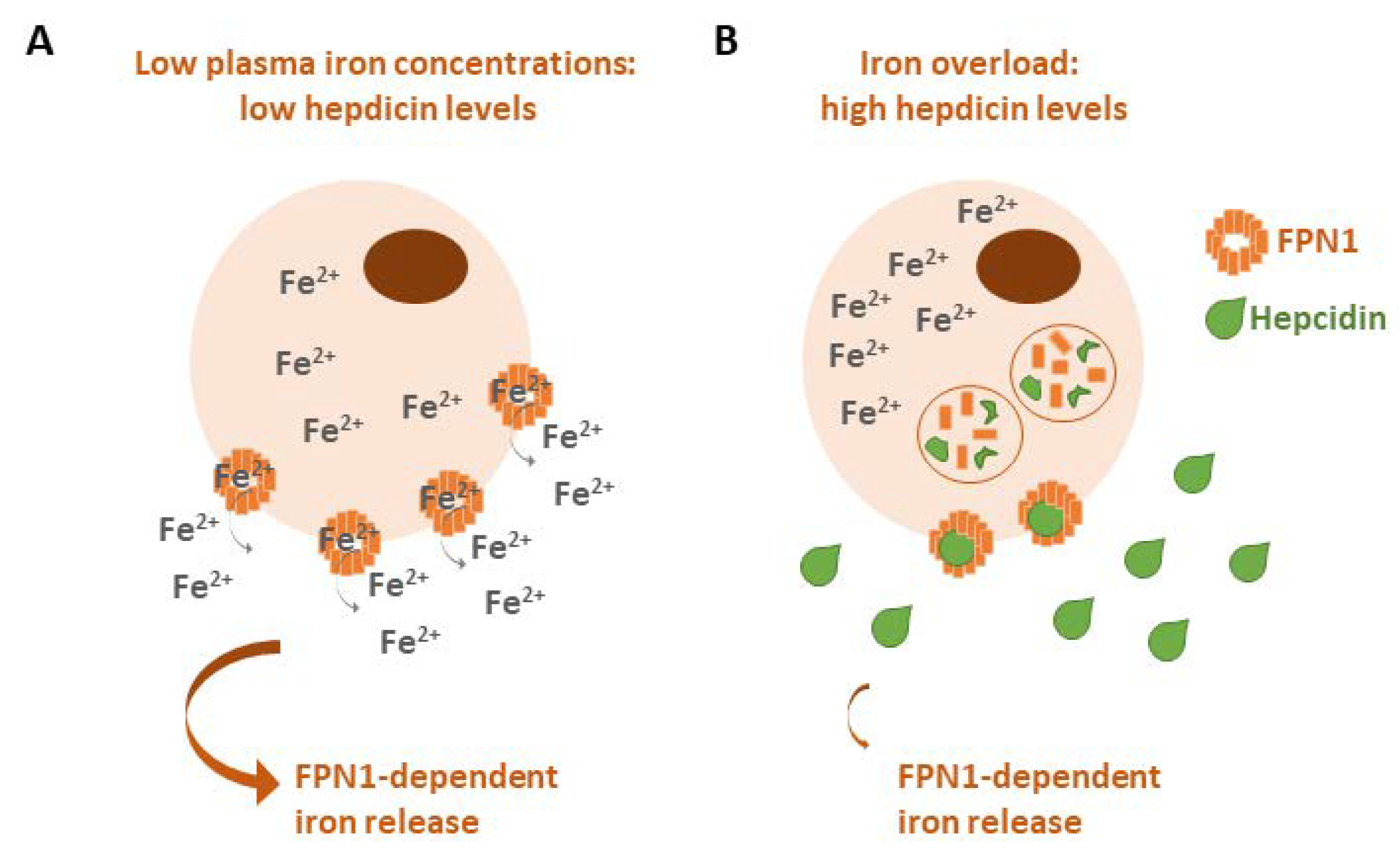
2.3. Ferroportin (FPN1)
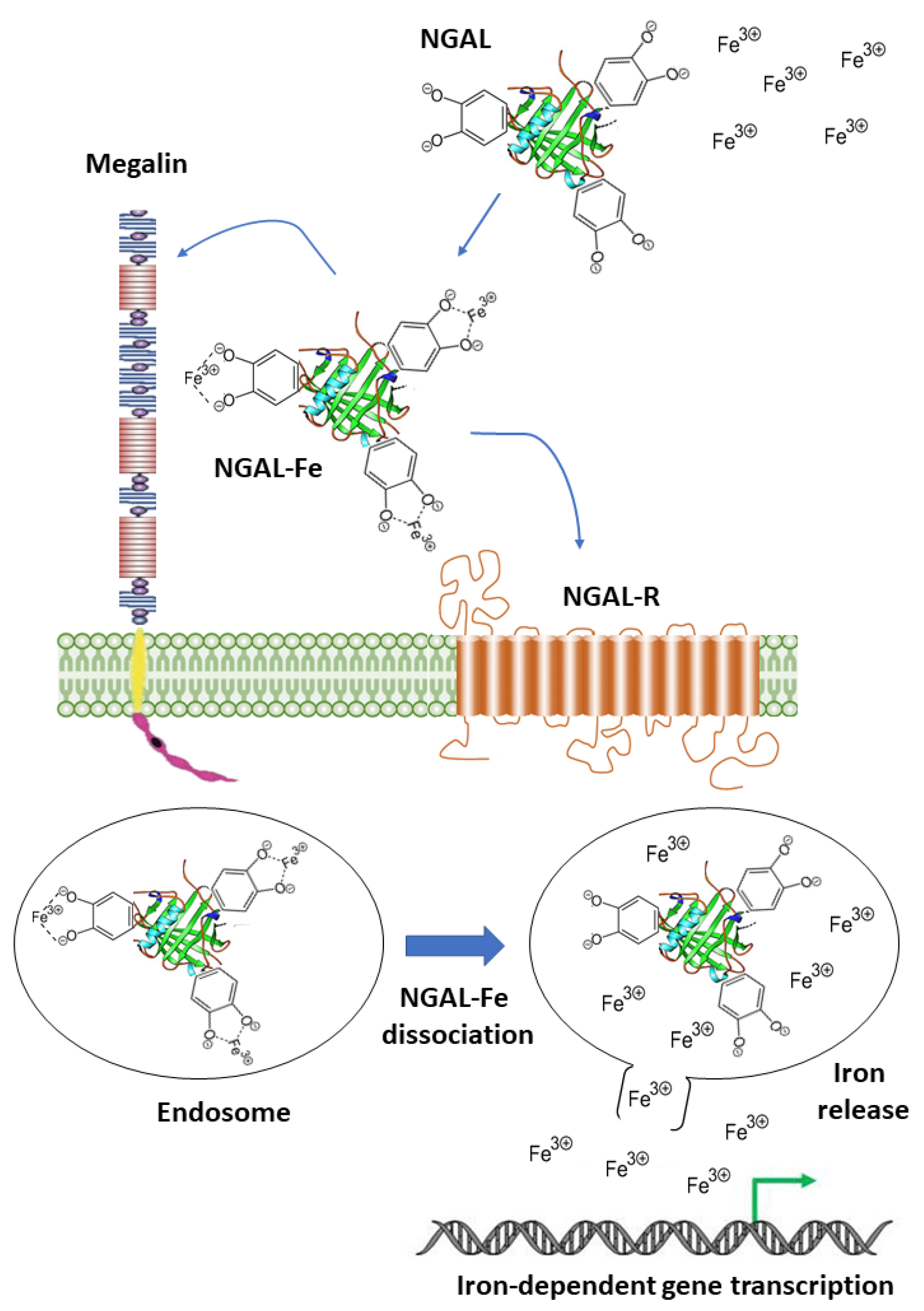
2.4. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
2.5. Other Iron Proteins
- Duodenal cytochrome B (DCYTB): a ferrireductase that reduces Fe3+ to Fe2+ to allow for the uptake of iron by DMT1 on the surfaces of duodenal cells [5]. Lemler and co-workers found that DCYTB expression is a favorable prognostic factor in breast cancer patients because it correlates with a better response to therapy and an increased progression-free survival [146]. Interestingly, they also showed that upregulated DCYTB improves outcomes for breast cancer patients via an iron-unrelated mechanism involving the inhibition of FAK activation and cell adhesion [146].
- Hepcidin: a small peptide produced by the liver that is able to induce FPN1 degradation to block iron export from cells [8]. Hepcidin is overexpressed in several human tumors, such as breast, lung and prostate cancers, as well as multiple myeloma, for its property to promote neoplastic growth by increasing iron retention in malignant cells [147]. In a breast tumor microenvironment, cancer-associated fibroblasts stimulated hepcidin expression in breast cancer cells via the production of IL-6 [148], while it has been found that hepcidin expression is associated with immune tumor infiltrates in lung cancer, particularly those constituting B cells, CD4 + T cells, macrophages, neutrophils and dendritic cells [149].
- Divalent metal transporter 1 (DMT1): a key protein in the regulation of iron homeostasis for its ability to enable the translocation of Fe2+ to the cytosol after iron endocytosis [5]. Blocking DMT1 in colorectal cancer has been shown to suppress cancer progression [26], and using DMT1 inhibitors has been reported to selectively kill iron-addicted cancer stem cells by inducing lysosomal iron overload [150]. Interestingly, DMT1 inhibition promotes ferroptosis in head and neck cancers [151].
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yin, N.H.; Louvat, P.; Thibault-DE-Chanvalon, A.; Sebilo, M.; Amouroux, D. Iron isotopic fractionation driven by low-temperature biogeochemical processes. Chemosphere 2023, 316, 137802. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Di Betto, G.; Bergamini, E.; Buzzetti, E.; Corradini, E.; Ventura, P. Iron Metabolism in the Disorders of Heme Biosynthesis. Metabolites 2022, 12, 819. [Google Scholar] [CrossRef]
- Khan, A.; Singh, P.; Srivastava, A. Iron: Key player in cancer and cell cycle? J. Trace Elem. Med. Biol. 2020, 62, 126582. [Google Scholar] [CrossRef] [PubMed]
- Basak, T.; Kanwar, R.K. Iron imbalance in cancer: Intersection of deficiency and overload. Cancer Med. 2022, 11, 3837–3853. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.K. Iron Regulatory Protein (IRP)-Iron Responsive Element (IRE) Signaling Pathway in Human Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. Int. Rev. J. 2017, 8, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Pantopoulos, K. Inherited Disorders of Iron Overload. Front. Nutr. 2018, 5, 103. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Ganz, T. Hepcidin and iron-loading anemias. Haematologica 2006, 91, 727–732. [Google Scholar]
- Ali, M.Y.; Oliva, C.R.; Flor, S.; Griguer, C.E. Mitoferrin, Cellular and Mitochondrial Iron Homeostasis. Cells 2022, 11, 3464. [Google Scholar] [CrossRef] [PubMed]
- Flo, T.H.; Smith, K.D.; Sato, S.; Rodriguez, D.J.; Holmes, M.A.; Strong, R.K.; Akira, S.; Aderem, A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 2004, 432, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, E.; Leonardi, A.; Pacifico, F. NGAL as a Potential Target in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 12333. [Google Scholar] [CrossRef]
- Yang, J.; Goetz, D.; Li, J.Y.; Wang, W.; Mori, K.; Setlik, D.; Du, T.; Erdjument-Bromage, H.; Tempst, P.; Strong, R.; et al. An iron delivery pathway mediated by a lipocalin. Mol. Cell 2002, 5, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Borregaard, N.; Kjeldsen, L.; Moses, M.A. The high molecular weight urinary matrix metalloproteinase (MMP) activity is a complex of gelatinase B/MMP-9 and neutrophil gelatinase-associated lipocalin (NGAL). Modulation of MMP-9 activity by NGAL. J. Biol. Chem. 2001, 276, 37258–37265. [Google Scholar] [CrossRef] [Green Version]
- Coradduzza, D.; Congiargiu, A.; Chen, Z.; Zinellu, A.; Carru, C.; Medici, S. Ferroptosis and Senescence: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 3658. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Fratta Pasini, A.M.; Stranieri, C.; Busti, F.; Di Leo, E.G.; Girelli, D.; Cominacini, L. New Insights into the Role of Ferroptosis in Cardiovascular Diseases. Cells 2023, 12, 867. [Google Scholar] [CrossRef]
- Xu, Y.; Zhao, J.; Zhao, Y.; Zhou, L.; Qiao, H.; Xu, Q.; Liu, Y. The role of ferroptosis in neurodegenerative diseases. Mol. Biol. Rep. 2023, 50, 1655–1661. [Google Scholar] [CrossRef]
- Chen, H.; Wang, C.; Liu, Z.; He, X.; Tang, W.; He, L.; Feng, Y.; Liu, D.; Yin, Y.; Li, T. Ferroptosis and Its Multifaceted Role in Cancer: Mechanisms and Therapeutic Approach. Antioxidants 2022, 11, 1504. [Google Scholar] [CrossRef]
- Hsu, M.Y.; Mina, E.; Roetto, A.; Porporato, P.E. Iron: An Essential Element of Cancer Metabolism. Cells 2020, 9, 2591. [Google Scholar] [CrossRef] [PubMed]
- Lelièvre, P.; Sancey, L.; Coll, J.L.; Deniaud, A.; Busser, B. Iron Dysregulation in Human Cancer: Altered Metabolism, Biomarkers for Diagnosis, Prognosis, Monitoring and Rationale for Therapy. Cancers 2020, 12, 3524. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and Cancer: 2020 Vision. Cancer Res. 2020, 80, 5435–5448. [Google Scholar] [CrossRef] [PubMed]
- Vela, D. Iron in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1259, 39–51. [Google Scholar] [CrossRef]
- Szymonik, J.; Wala, K.; Górnicki, T.; Saczko, J.; Pencakowski, B.; Kulbacka, J. The Impact of Iron Chelators on the Biology of Cancer Stem Cells. Int. J. Mol. Sci. 2021, 23, 89. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogeneic RAS and the p53 tumor suppressor. PLos Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M.; et al. Tumour biology: Senescence in premalignant tumours. Nature 2005, 436, 642. [Google Scholar] [CrossRef] [PubMed]
- Cichowski, K.; Hahn, W.C. Unexpected pieces to the senescence puzzle. Cell 2008, 133, 958–961. [Google Scholar] [CrossRef] [Green Version]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar]
- Mirzayans, R.; Scott, A.; Cameron, M.; Murray, D. Induction of accelerated senescence by gamma radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiat. Res. 2005, 163, 53–62. [Google Scholar] [CrossRef]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Campisi, J. The metabolic roots of senescence: Mechanisms and opportunities for intervention. Nat. Metab. 2021, 3, 1290–1301. [Google Scholar] [CrossRef]
- Kwon, S.M.; Hong, S.M.; Lee, Y.K.; Min, S.; Yoon, G. Metabolic features and regulation in cell senescence. BMB Rep. 2019, 52, 5–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Pan, K.H.; Cohen, S.N. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc. Natl. Acad. Sci. USA 2003, 100, 3251–3256. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killilea, D.W.; Wong, S.L.; Cahaya, H.S.; Atamna, H.; Ames, B.N. Iron accumulation during cellular senescence. Ann. N. Y. Acad. Sci. 2004, 1019, 365–367. [Google Scholar] [CrossRef]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef]
- Tian, Y.; Tian, Y.; Yuan, Z.; Zeng, Y.; Wang, S.; Fan, X.; Yang, D.; Yang, M. Iron Metabolism in Aging and Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 3612. [Google Scholar] [CrossRef]
- Chen, W.J.; Kung, G.P.; Gnana-Prakasam, J.P. Role of Iron in Aging Related Diseases. Antioxidants 2022, 11, 865. [Google Scholar] [CrossRef]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar]
- Panaccio, M.; Zalcberg, J.R.; Thompson, C.H.; Leyden, M.J.; Sullivan, J.R.; Lichtenstein, M.; McKenzie, I.F. Heterogeneity of the human transferrin receptor and use of anti-transferrin receptor antibodies to detect tumours in vivo. Immunol. Cell Biol. 1987, 65, 461–472. [Google Scholar] [CrossRef]
- Essaghir, A.; Demoulin, J.B. A minimal connected network of transcription factors regulated in human tumors and its application to the quest for universal cancer biomarkers. PLoS ONE 2012, 7, e39666. [Google Scholar] [CrossRef]
- Guo, Q.; Li, L.; Hou, S.; Yuan, Z.; Li, C.; Zhang, W.; Zheng, L.; Li, X. The Role of Iron in Cancer Progression. Front. Oncol. 2021, 11, 778492. [Google Scholar] [CrossRef]
- O’Donnell, K.A.; Yu, D.; Zeller, K.I.; Kim, J.W.; Racke, F.; Thomas-Tikhonenko, A.; Dang, C.V. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol. Cell. Biol. 2006, 26, 2373–2386. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, L.; Tacchini, L.; Cairo, G. HIF-1-mediated activation of transferrin receptor gene transcription by iron chelation. Nucleic Acids Res. 1999, 27, 4223–4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Jian, J.; Bosland, M.; Frenkel, K.; Bernhardt, G.; Huang, X. Roles of hormone replacement therapy and iron in proliferation of breast epithelial cells with different estrogen and progesterone receptor status. Breast 2008, 17, 172–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.P.; Elliott, R.L. Decreased iron in cancer cells and their microenvironment improves cytolysis of breast cancer cells by natural killer cells. Anticancer Res. 2017, 37, 2297–2305. [Google Scholar] [CrossRef] [Green Version]
- Kenneth, N.S.; Mudie, S.; Naron, S.; Rocha, S. TfR1 interacts with the IKK complex and is involved in IKK-NF-κB signalling. Biochem. J. 2013, 449, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Fan, Y.; Hou, J.; Liu, B.; Zhang, B.; Shang, Y.; Chang, Y.; Cao, P.; Tan, K. Integrated analysis identifies TfR1 as a prognostic biomarker which correlates with immune infiltration in breast cancer. Aging 2021, 13, 21671–21699. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; He, H.; Huang, J.; Wang, C.; Dong, Y.; Lin, R.; Cheng, Z.; Qiu, Q.; Hong, L. Identification and validation of transferrin receptor protein 1 for predicting prognosis and immune infiltration in lower grade glioma. Front. Mol. Neurosci. 2022, 15, 972308. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Fu, X.; Wang, Y.; Liu, H.; Jiang, Y.; Zhao, Z.; You, F. Transferrin receptor regulates malignancies and the stemness of hepatocellular carcinoma-derived cancer stem-like cells by affecting iron accumulation. PLoS ONE 2020, 15, e0243812. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Bernabeu, E.; Rodriguez, J.A.; Patel, S.; Kozman, M.; Chiappetta, D.A.; Holler, E.; Ljubimova, J.Y.; Helguera, G.; Penichet, M.L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820, 291–317. [Google Scholar] [CrossRef] [Green Version]
- Candelaria, P.V.; Leoh, L.S.; Penichet, M.L.; Daniels-Wells, T.R. Antibodies Targeting the Transferrin Receptor 1 (TfR1) as Direct Anti-cancer Agents. Front. Immunol. 2021, 12, 607692. [Google Scholar] [CrossRef] [PubMed]
- Calzolari, A.; Larocca, L.M.; Deaglio, S.; Finisguerra, V.; Boe, A.; Raggi, C.; Ricci-Vitani, L.; Pierconti, F.; Malavasi, F.; De Maria, R.; et al. Transferrin receptor 2 is frequently and highly expressed in glioblastomas. Transl. Oncol. 2010, 3, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calzolari, A.; Oliviero, I.; Deaglio, S.; Mariani, G.; Biffoni, M.; Sposi, N.M.; Malavasi, F.; Peschle, C.; Testa, U. Transferrin receptor 2 is frequently expressed in human cancer cell lines. Blood Cells Mol. Dis. 2007, 39, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Calzolari, A.; Finisguerra, V.; Oliviero, I.; Deaglio, S.; Mariani, G.; Malavasi, F.; Testa, U. Regulation of transferrin receptor 2 in human cancer cell lines. Blood Cells Mol. Dis. 2009, 42, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Voth, B.; Nagasawa, D.T.; Pelargos, P.E.; Chung, L.K.; Ung, N.; Gopen, Q.; Tenn, S.; Kamei, D.T.; Yang, I. Transferrin receptors and glioblastoma multiforme: Current findings and potential for treatment. J. Clin. Neurosci. 2015, 22, 1071–1076. [Google Scholar] [CrossRef]
- Pascal, T.; Debacq-Chainiaux, F.; Chrétien, A.; Bastin, C.; Dabée, A.F.; Bertholet, V.; Remacle, J.; Toussaint, O. Comparison of replicative senescence and stress-induced premature senescence combining differential display and low-density DNA arrays. FEBS Lett. 2005, 579, 3651–3659. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.R.; Fenwick, P.S.; Donnelly, L.E.; Barnes, P.J.; Cloonan, S.M. Altered iron metabolism and elevated cellular senescence in COPD small airway epithelial cells. Eur. Respir. J. 2020, 56, 3707. [Google Scholar]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell. Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Admasu, T.D.; Kim, K.; Rae, M.; Avelar, R.; Gonciarz, R.L.; Rebbaa, A.; Pedro de Magalhães, J.; Renslo, A.R.; Stolzing, A.; Sharma, A. Selective ablation of primary and paracrine senescent cells by targeting iron dyshomeostasis. Cell. Rep. 2023, 42, 112058. [Google Scholar] [CrossRef]
- Jin, Y.; Zhao, L.; Wang, S.; Zhang, X.; Quan, J.; Lin, Z.; Piao, J. RSL1D1 knockdown induces ferroptosis and mediates ferrous iron accumulation in senescent cells by inhibiting FTH1 mRNA stability. Carcinogenesis 2023, 44, 129–142. [Google Scholar] [CrossRef]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, A.A.; Han, B.; Connor, J.R. Ferritin stimulates breast cancer cells through an iron independent mechanism and is localized within tumor-associated macrophages. Breast Cancer Res. Treat. 2013, 137, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zhao, H.; Luo, C.; Lei, T.; Zhang, M. Knockdown of ferritin heavy chain (FTH) inhibits the migration of prostate cancer through reducing S100A4, S100A2, and S100P expression. Transl. Cancer Res. 2020, 9, 5418–5429. [Google Scholar] [CrossRef]
- Schonberg, D.L.; Miller, T.E.; Wu, Q.; Flavahan, W.A.; Das, N.K.; Hale, J.S.; Hubert, C.G.; Mack, S.C.; Jarrar, A.M.; Karl, R.T.; et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 2015, 28, 441–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Zhou, C.; Jing, Q.; Li, Y.; Yang, J.; Yang, C.; Wang, L.; Hu, J.; Li, H.; Wang, H.; et al. FTH promotes the proliferation and renders the HCC cells specifically resist to ferroptosis by maintaining iron homeostasis. Cancer Cell Int. 2021, 21, 709. [Google Scholar] [CrossRef]
- Wu, T.; Li, Y.; Liu, B.; Zhang, S.; Wu, L.; Zhu, X.; Chen, Q. Expression of Ferritin Light Chain (FTL) Is Elevated in Glioblastoma, and FTL Silencing Inhibits Glioblastoma Cell Proliferation via the GADD45/JNK Pathway. PLoS ONE 2016, 11, e0149361. [Google Scholar] [CrossRef] [Green Version]
- Kambara, T.; Amatya, V.J.; Kushitani, K.; Fujii, Y.; Endo, I.; Takeshima, Y. Downregulation of FTL decreases proliferation of malignant mesothelioma cells by inducing G1 cell cycle arrest. Oncol. Lett. 2022, 23, 174. [Google Scholar] [CrossRef]
- Cui, Z.; Li, W.; Wang, Y.; Zhao, M.; Liu, K.; Yang, Y.; Teng, S.; Zhang, N.; Min, L.; Li, P.; et al. M2 Macrophage-Derived Exosomal Ferritin Heavy Chain Promotes Colon Cancer Cell Proliferation. Biol. Trace Elem. Res. 2022, 201, 3717–3728. [Google Scholar] [CrossRef]
- Alkhateeb, A.A.; Connor, J.R. The significance of ferritin in cancer: Anti-oxidation, inflammation and tumorigenesis. Biochim. Biophys. Acta 2013, 1836, 245–254. [Google Scholar] [CrossRef]
- Biamonte, F.; Battaglia, A.M.; Zolea, F.; Oliveira, D.M.; Aversa, I.; Santamaria, G.; Giovannone, E.D.; Rocco, G.; Viglietto, G.; Costanzo, F. Ferritin heavy subunit enhances apoptosis of non-small cell lung cancer cells through modulation of miR-125b/p53 axis. Cell Death Dis. 2018, 9, 1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santambrogio, P.; Erba, B.G.; Campanella, A.; Cozzi, A.; Causarano, V.; Cremonesi, L.; Gallì, A.; Della Porta, M.G.; Invernizzi, R.; Levi, S. Overexpression of mitochondrial ferritin affects the JAK2/STAT5 pathway in K562 cells and causes mitochondrial iron accumulation. Haematologica 2011, 96, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.C.; Jiang, X.; Elliott, R.L.; Head, J.F. Antisense ferritin oligonucleotides inhibit growth and induce apoptosis in human breast carcinoma cells. Anticancer Res. 2002, 22, 1513–1524. [Google Scholar] [PubMed]
- Cozzi, A.; Levi, S.; Corsi, B.; Santambrogio, P.; Campanella, A.; Gerardi, G.; Arosio, P. Role of iron and ferritin in TNFα-induced apoptosis in HeLa cells. FEBS Lett. 2003, 537, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Baldi, A.; Lombardi, D.; Russo, P.; Palescandolo, E.; De Luca, A.; Santini, D.; Baldi, F.; Rossiello, L.; Dell’Anna, M.L.; Mastrofrancesco, A.; et al. Ferritin Contributes to Melanoma Progression by Modulating Cell Growth and Sensitivity to Oxidative Stress. Clin. Cancer Res. 2005, 11, 3175–3183. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.M.; Fitch, M.E.; Yuan, M.; Parslow, T.G.; Shu, H.G. Bax can associate with ferritin heavy chain (FHC) resulting in inhibition of bax-mediated apoptosis. Int. J. Radiat. Oncol. 2001, 51, 189. [Google Scholar] [CrossRef]
- Sioutas, A.; Vainikka, L.K.; Kentson, M.; Dam-Larsen, S.; Wennerström, U.; Jacobson, P.; Persson, H.L. Oxidant-induced autophagy and ferritin degradation contribute to epithelial–mesenchymal transition through lysosomal iron. J. Inflamm. Res. 2017, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Aversa, I.; Zolea, F.; Ieranò, C.; Bulotta, S.; Trotta, A.M.; Faniello, M.C.; De Marco, C.; Malanga, D.; Biamonte, F.; Viglietto, G.; et al. Epithelial-to-mesenchymal transition in FHC-silenced cells: The role of CXCR4/CXCL12 axis. J. Exp. Clin. Cancer Res. 2017, 36, 104. [Google Scholar] [CrossRef] [Green Version]
- Eckard, J.; Dai, J.; Wu, J.; Jian, J.; Yang, Q.; Chen, H.; Costa, M.; Frenkel, K.; Huang, X. Effects of cellular iron deficiency on the formation of vascular endothelial growth factor and angiogenesis. Iron deficiency and angiogenesis. Cancer Cell Int. 2010, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Mi, S.; Du, J.; Liu, J.; Hou, K.; Ji, H.; Ma, S.; Ba, Y.; Chen, L.; Xie, R.; Hu, S. FtMt promotes glioma tumorigenesis and angiogenesis via lncRNA SNHG1/miR-9-5p axis. Cell. Signal. 2020, 75, 109749. [Google Scholar] [CrossRef]
- Raggi, C.; Gammella, E.; Correnti, M.; Buratti, P.; Forti, E.; Andersen, J.B.; Alpini, G.; Glaser, S.; Alvaro, D.; Invernizzi, P.; et al. Dysregulation of Iron Metabolism in Cholangiocarcinoma Stem-like Cells. Sci. Rep. 2017, 7, 17667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanvorachote, P.; Luanpitpong, S. Iron induces cancer stem cells and aggressive phenotypes in human lung cancer cells . Am. J. Physiol. Cell Physiol. 2016, 310, C728–C739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rychtarcikova, Z.; Lettlova, S.; Tomkova, V.; Korenkova, V.; Langerova, L.; Simonova, E.; Zjablovskaja, P.; Alberich-Jorda, M.; Neuzil, J.; Truksa, J. Tumor initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017, 8, 6376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.; Girotti, A.W. Elevated ferritin production, iron containment, and oxidant resistance in hemin-treated leukemia cells. Arch. Biochem. Biophys. 1997, 346, 131–141. [Google Scholar] [CrossRef]
- Wu, J.; Liu, H.; Zhang, G.; Gu, L.; Zhang, Y.; Gao, J.; Wei, Y.; Ma, Z. Antileukemia Effect of Ciclopirox Olamine Is Mediated by Downregulation of Intracellular Ferritin and Inhibition β-Catenin-c-Myc Signaling Pathway in Glucocorticoid Resistant T-ALL Cell Lines. PLoS ONE 2016, 11, e0161509. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Madhankumar, A.B.; Slagle-Webb, B.; Sheehan, J.M.; Surguladze, N.; Connor, J.R. Heavy chain ferritin siRNA delivered by cationic liposomes increases sensitivity of cancer cells to chemotherapeutic agents. Cancer Res. 2011, 71, 2240–2249. [Google Scholar] [CrossRef] [Green Version]
- Connor, J.R. Role of H-Ferritin in Radiosensitivity of Human Glioma Cells. Cancer Biol. Treat. 2016, 3, e0221952. [Google Scholar] [CrossRef]
- Chekhun, V.F.; Lukyanova, N.Y.; Burlaka, A.P.; Bezdenezhnykh, N.A.; Shpyleva, S.I.; Tryndyak, V.P.; Beland, F.A.; Pogribny, I.P. Iron metabolism disturbances in the MCF-7 human breast cancer cells with acquired resistance to doxorubicin and cisplatin. Int. J. Oncol. 2013, 43, 1481–1486. [Google Scholar] [CrossRef] [Green Version]
- Tirinato, L.; Marafioti, M.G.; Pagliari, F.; Jansen, J.; Aversa, I.; Hanley, R.; Nisticò, C.; Garcia-Calderón, D.; Genard, G.; Guerreiro, J.F.; et al. Lipid droplets and ferritin heavy chain: A devilish liaison in human cancer cell radioresistance. Elife 2021, 10, e72943. [Google Scholar] [CrossRef]
- Ott, C.; König, J.; Höhn, A.; Jung, T.; Grune, T. Reduced autophagy leads to an impaired ferritin turnover in senescent fibroblasts. Free Radic. Biol. Med. 2016, 101, 325–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovira, M.; Sereda, R.; Pladevall-Morera, D.; Ramponi, V.; Marin, I.; Maus, M.; Madrigal-Matute, J.; Díaz, A.; García, F.; Muñoz, J.; et al. The lysosomal proteome of senescent cells contributes to the senescence secretome. Aging Cell 2022, 21, e13707. [Google Scholar] [CrossRef] [PubMed]
- Robbins, E.; Levine, E.M.; Eagle, H. Morphologic changes accompanying senescence of cultured human diploid cells. J. Exp. Med. 1970, 131, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Estévez-Souto, V.; Da Silva-Álvarez, S.; Collado, M. The role of extracellular vesicles in cellular senescence. FEBS J. 2023, 290, 1203–1211. [Google Scholar] [CrossRef]
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef]
- Go, S.; Kang, M.; Kwon, S.P.; Jung, M.; Jeon, O.H.; Kim, B.S. The Senolytic Drug JQ1 Removes Senescent Cells via Ferroptosis. Tissue Eng. Regen. Med. 2021, 18, 841–850. [Google Scholar] [CrossRef]
- Cozzi, A.; Orellana, D.I.; Santambrogio, P.; Rubio, A.; Cancellieri, C.; Giannelli, S.; Ripamonti, M.; Taverna, S.; Di Lullo, G.; Rovida, E.; et al. Stem Cell Modeling of Neuroferritinopathy Reveals Iron as a Determinant of Senescence and Ferroptosis during Neuronal Aging. Stem Cell Rep. 2019, 13, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Tesfay, L.; Clausen, K.A.; Kim, J.W.; Hegde, P.; Wang, X.; Miller, L.D.; Deng, Z.; Blanchette, N.; Arvedson, T.; Miranti, C.K.; et al. Hepcidin Regulation in Prostate and Its Disruption in Prostate Cancer. Cancer Res. 2015, 75, 2254–2263. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Lu, Y.; Cheng, X.; Wang, J. Hepcidin and Ferroportin Expression in Breast Cancer Tissue and Serum and Their Relationship With Anemia. Curr. Oncol. 2016, 23, e24–e26. [Google Scholar] [CrossRef] [Green Version]
- Babu, K.R.; Muckenthaler, M.U. miR-20a Regulates Expression of the Iron Exporter Ferroportin in Lung Cancer. J. Mol. Med. 2016, 94, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and Iron Regulation in Breast Cancer Progression and Prognosis. Sci. Trans. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef] [PubMed]
- Sornjai, W.; Nguyen Van Long, F.; Pion, N.; Pasquer, A.; Saurin, J.C.; Marcel, V.; Diaz, J.J.; Mertani, H.C.; Smith, D.R. Iron and Hepcidin Mediate Human Colorectal Cancer Cell Growth. Chem.-Biol. Interact. 2020, 319, 109021. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Manz, D.H.; Torti, S.V.; Torti, F.M. Effects of Ferroportin-Mediated Iron Depletion in Cells Representative of Different Histological Subtypes of Prostate Cancer. Antioxid. Redox Signal. 2019, 30, 1043–1061. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, S.; Chen, Y.; Zhang, D.; Yuan, L.; Cong, H.; Liu, S. An Important Role of the Hepcidin-Ferroportin Signaling in Affecting Tumor Growth and Metastasis. Acta Biochim. Biophys. Sin. 2015, 47, 703–715. [Google Scholar] [CrossRef] [Green Version]
- Shan, Z.; Wei, Z.; Shaikh, Z.A. Suppression of Ferroportin Expression by Cadmium Stimulates Proliferation, EMT, and Migration in Triple-Negative Breast Cancer Cells. Toxicol. Appl. Pharmacol. 2018, 356, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, J.; Feng, J.; Wang, J. E4BP4 Promotes Thyroid Cancer Proliferation by Modulating Iron Homeostasis Through Repression of Hepcidin. Cell Death Dis. 2018, 9, 987. [Google Scholar] [CrossRef] [Green Version]
- Geng, N.; Shi, B.J.; Li, S.L.; Zhong, Z.Y.; Li, Y.C.; Xua, W.L.; Zhou, H.; Cai, J.H. Knockdown of Ferroportin Accelerates Erastin-Induced Ferroptosis in Neuroblastoma Cells. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3826–3836. [Google Scholar] [CrossRef]
- Tang, Z.; Jiang, W.; Mao, M.; Zhao, J.; Chen, J.; Cheng, N. Deubiquitinase USP35 Modulates Ferroptosis in Lung Cancer via Targeting Ferroportin. Clin. Trans. Med. 2021, 11, e390. [Google Scholar] [CrossRef]
- Belvin, B.R.; Lewis, J.P. Ferroportin depletes iron needed for cell cycle progression in head and neck squamous cell carcinoma. Front. Oncol. 2023, 12, 1025434. [Google Scholar] [CrossRef]
- Jiang, H.; Muir, R.K.; Gonciarz, R.L.; Olshen, A.B.; Yeh, I.; Hann, B.C.; Zhao, N.; Wang, Y.H.; Behr, S.C.; Korkola, J.E.; et al. Ferrous iron-activatable drug conjugate achieves potent MAPK blockade in KRAS-driven tumors. J. Exp. Med. 2022, 219, e20210739. [Google Scholar] [CrossRef]
- Bauer, M.; Eickhoff, J.C.; Gould, M.N.; Mundhenke, C.; Maass, N.; Friedl, A. Neutrophil gelatinase-associated lipocalin (NGAL) is a predictor of poor prognosis in human primary breast cancer. Breast Cancer Res. Treat. 2008, 108, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.T.; Aigner, F.; Trenkwalder, B.; Zitt, M.; Vallant, N.; Perathoner, A.; Margreiter, C.; Moser, P.; Pratschke, J.; Amberger, A. Up-regulation of neutrophil gelatinase-associated lipocalin in colorectal cancer predicts poor patient survival. World J. Surg. 2014, 38, 2160–2167. [Google Scholar] [CrossRef]
- Villalva, C.; Sorel, N.; Bonnet, M.L.; Guilhot, J.; Mayeur-Rousse, C.; Guilhot, F.; Chomel, J.C.; Turhan, A.G. Neutrophil gelatinase-associated lipocalin expression in chronic myeloid leukemia. Leuk. Lymphoma 2008, 49, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Bauvois, B.; Pramil, E.; Jondreville, L.; Chapiro, E.; Quiney, C.; Maloum, K.; Susin, S.A.; Nguyen-Khac, F. Relation of Neutrophil Gelatinase-Associated Lipocalin Overexpression to the Resistance to Apoptosis of Tumor B Cells in Chronic Lymphocytic Leukemia. Cancers 2020, 12, 2124. [Google Scholar] [CrossRef] [PubMed]
- Moniaux, N.; Chakraborty, S.; Yalniz, M.; Gonzalez, J.; Shostrom, V.K.; Standop, J.; Lele, S.M.; Ouellette, M.; Pour, P.M.; Sasson, A.R.; et al. Early diagnosis of pancreatic cancer: Neutrophil gelatinase-associated lipocalin as a marker of pancreatic intraepithelial neoplasia. Br. J. Cancer 2008, 98, 1540–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannetti, A.; Pacifico, F.; Acquaviva, R.; Lavorgna, A.; Crescenzi, E.; Vascotto, C.; Tell, G.; Salzano, A.M.; Scaloni, A.; Vuttariello, E.; et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc. Natl. Acad. Sci. USA 2008, 105, 14058–14063. [Google Scholar] [CrossRef]
- Miyamoto, T.; Kashima, H.; Yamada, Y.; Kobara, H.; Asaka, R.; Ando, H.; Higuchi, S.; Ida, K.; Mvunta, D.H.; Shiozawa, T. Lipocalin 2 Enhances Migration and Resistance against Cisplatin in Endometrial Carcinoma Cells. PLoS ONE 2016, 11, e0155220. [Google Scholar] [CrossRef] [Green Version]
- Shiiba, M.; Saito, K.; Fushimi, K.; Ishigami, T.; Shinozuka, K.; Nakashima, D.; Kouzu, Y.; Koike, H.; Kasamatsu, A.; Sakamoto, Y.; et al. Lipocalin-2 is associated with radioresistance in oral cancer and lung cancer cells. Int. J. Oncol. 2013, 42, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Tung, M.C.; Hsieh, S.C.; Yang, S.F.; Cheng, C.W.; Tsai, R.T.; Wang, S.C.; Huang, M.H.; Hsieh, Y.H. Knockdown of lipocalin-2 suppresses the growth and invasion of prostate cancer cells. Prostate 2013, 73, 1281–1290. [Google Scholar] [CrossRef]
- Fernàndez, C.A.; Yan, L.; Louis, G.; Yang, J.; Kutok, J.L.; Moses, M.A. The matrix metalloproteinase-9/neutrophil gelatinase-associated lipocalin complex plays a role in breast tumor growth and is present in the urine of breast cancer patients. Clin. Cancer Res. 2005, 11, 5390–5395. [Google Scholar] [CrossRef] [Green Version]
- Kubben, F.J.; Sier, C.F.; Hawinkels, L.J.; Tschesche, H.; van Duijn, W.; Zuidwijk, K.; van der Reijden, J.J.; Hanemaaijer, R.; Griffioen, G.; Lamers, C.B.; et al. Clinical evidence for a protective role of lipocalin-2 against MMP-9 autodegradation and the impact for gastric cancer. Eur. J. Cancer 2007, 43, 1869–1876. [Google Scholar] [CrossRef]
- Srdelić Mihalj, S.; Kuzmić-Prusac, I.; Zekić-Tomaš, S.; Šamija-Projić, I.; Čapkun, V. Lipocalin-2 and matrix metalloproteinase-9 expression in high-grade endometrial cancer and their prognostic value. Histopathology 2015, 67, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.K.; Pinoé-Schmidt, M.; Weiskirchen, R. Lipocalin-2 (LCN2) Deficiency Leads to Cellular Changes in Highly Metastatic Human Prostate Cancer Cell Line PC-3. Cells 2022, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Volpe, V.; Raia, Z.; Sanguigno, L.; Somma, D.; Mastrovito, P.; Moscato, F.; Mellone, S.; Leonardi, A.; Pacifico, F. NGAL controls the metastatic potential of anaplastic thyroid carcinoma cells. J. Clin. Endocrinol. Metab. 2013, 98, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Sharma, S.; Wu, K.; Wu, S.Y.; Xing, F.; Liu, Y.; Zhao, D.; Deshpande, R.P.; D’Agostino, R.B., Jr.; Watabe, K. Nicotine promotes breast cancer metastasis by stimulating N2 neutrophils and generating pre-metastatic niche in lung. Nat. Commun. 2021, 12, 474. [Google Scholar] [CrossRef]
- Ören, B.; Urosevic, J.; Mertens, C.; Mora, J.; Guiu, M.; Gomis, R.R.; Weigert, A.; Schmid, T.; Grein, S.; Brüne, B.; et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J. Pathol. 2016, 239, 274–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacifico, F.; Pisa, L.; Mellone, S.; Cillo, M.; Lepore, A.; Leonardi, A. NGAL promotes recruitment of tumor infiltrating leukocytes. Oncotarget 2018, 9, 30761–30772. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, N.; Choudhary, B.S.; Shah, S.G.; Khapare, N.; Dwivedi, N.; Gaikwad, A.; Joshi, N.; Raichanna, J.; Basu, S.; Gurjar, M.; et al. Lipocalin 2 expression promotes tumor progression and therapy resistance by inhibiting ferroptosis in colorectal cancer. Int. J. Cancer 2021, 149, 1495–1511. [Google Scholar] [CrossRef]
- Liu, J.; Song, X.; Kuang, F.; Zhang, Q.; Xie, Y.; Kang, R.; Kroemer, G.; Tang, D. NUPR1 is a critical repressor of ferroptosis. Nat. Commun. 2021, 12, 647. [Google Scholar] [CrossRef]
- Meier, J.K.; Schnetz, M.; Beck, S.; Schmid, T.; Dominguez, M.; Kalinovic, S.; Daiber, A.; Brüne, B.; Jung, M. Iron-Bound Lipocalin-2 Protects Renal Cell Carcinoma from Ferroptosis. Metabolites 2021, 11, 329. [Google Scholar] [CrossRef]
- Bahmani, B.; Roudkenar, M.H.; Halabian, R.; Jahanian-Najafabadi, A.; Amiri, F.; Jalili, M.A. Lipocalin 2 decreases senescence of bone marrow-derived mesenchymal stem cells under sub-lethal doses of oxidative stress. Cell Stress Chaperones 2014, 19, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin. Colorectal Cancer 2016, 15, 170–178.e3. [Google Scholar] [CrossRef] [PubMed]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef] [PubMed]
- Morales-Valencia, J.; Lau, L.; Martí-Nin, T.; Ozerdem, U.; David, G. Therapy-induced senescence promotes breast cancer cells plasticity by inducing Lipocalin-2 expression. Oncogene 2022, 41, 4361–4370. [Google Scholar] [CrossRef]
- Paramos-de-Carvalho, D.; Martins, I.; Cristóvão, A.M.; Dias, A.F.; Neves-Silva, D.; Pereira, T.; Chapela, D.; Farinho, A.; Jacinto, A.; Saúde, L. Targeting senescent cells improves functional recovery after spinal cord injury. Cell Rep. 2021, 36, 109334. [Google Scholar] [CrossRef]
- Lemler, D.J.; Lynch, M.L.; Tesfay, L.; Deng, Z.; Paul, B.T.; Wang, X.; Hegde, P.; Manz, D.H.; Torti, S.V.; Torti, F.M. DCYTB is a predictor of outcome in breast cancer that functions via iron-independent mechanisms. Breast Cancer Res. 2017, 19, 25. [Google Scholar] [CrossRef]
- Lin, F.; Tuffour, A.; Hao, G.; Peprah, F.A.; Huang, A.; Zhou, Y.; Zhang, H. Distinctive modulation of hepcidin in cancer and its therapeutic relevance. Front. Oncol. 2023, 13, 1141603. [Google Scholar] [CrossRef]
- Blanchette-Farra, N.; Kita, D.; Konstorum, A.; Tesfay, L.; Lemler, D.; Hegde, P.; Claffey, K.P.; Torti, F.M.; Torti, S.V. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene 2018, 37, 4013–4032. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, B.; Chen, F.; Song, Z.; Han, B.; Meng, Y.; Hou, J.; Cao, P.; Chang, Y.; Tan, K. Hepcidin upregulation in lung cancer: A potential therapeutic target associated with immune infiltration. Front. Immunol. 2021, 12, 612144. [Google Scholar] [CrossRef]
- Turcu, A.L.; Versini, A.; Khene, N.; Gaillet, C.; Cañeque, T.; Müller, S.; Rodriguez, R. DMT1 inhibitors kill cancer stem cells by blocking lysosomal iron translocation. Chemistry 2020, 26, 7369–7373. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.L. Promotion of ferroptosis in head and neck cancer with divalent metal transporter 1 inhibition or salinomycin. Hum. Cell 2023, 36, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, P.; Duan, X.; Cheng, M.; Xu, L.X. Deferoxamine-induced high expression of TfR1 and DMT1 enhanced iron uptake in triple-negative breast cancer cells by activating IL-6/PI3K/AKT pathway. OncoTargets Ther. 2019, 12, 4359–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, M.; Xue, X. Targeting iron metabolism in cancer therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crescenzi, E.; Leonardi, A.; Pacifico, F. Iron Metabolism in Cancer and Senescence: A Cellular Perspective. Biology 2023, 12, 989. https://doi.org/10.3390/biology12070989
Crescenzi E, Leonardi A, Pacifico F. Iron Metabolism in Cancer and Senescence: A Cellular Perspective. Biology. 2023; 12(7):989. https://doi.org/10.3390/biology12070989
Chicago/Turabian StyleCrescenzi, Elvira, Antonio Leonardi, and Francesco Pacifico. 2023. "Iron Metabolism in Cancer and Senescence: A Cellular Perspective" Biology 12, no. 7: 989. https://doi.org/10.3390/biology12070989
APA StyleCrescenzi, E., Leonardi, A., & Pacifico, F. (2023). Iron Metabolism in Cancer and Senescence: A Cellular Perspective. Biology, 12(7), 989. https://doi.org/10.3390/biology12070989