
Seasonality Is the Main Determinant of Microbial Diversity Associated to Snow/Ice around Concordia Station on the Antarctic Polar Plateau
 , , , ,
, , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. DNA Extraction and Amplicon Sequencing
2.3. Bioinformatics
2.4. Biodiversity Indexes and Statistical Analyses
3. Results
3.1. Bioinformatic Analysis of Raw Data
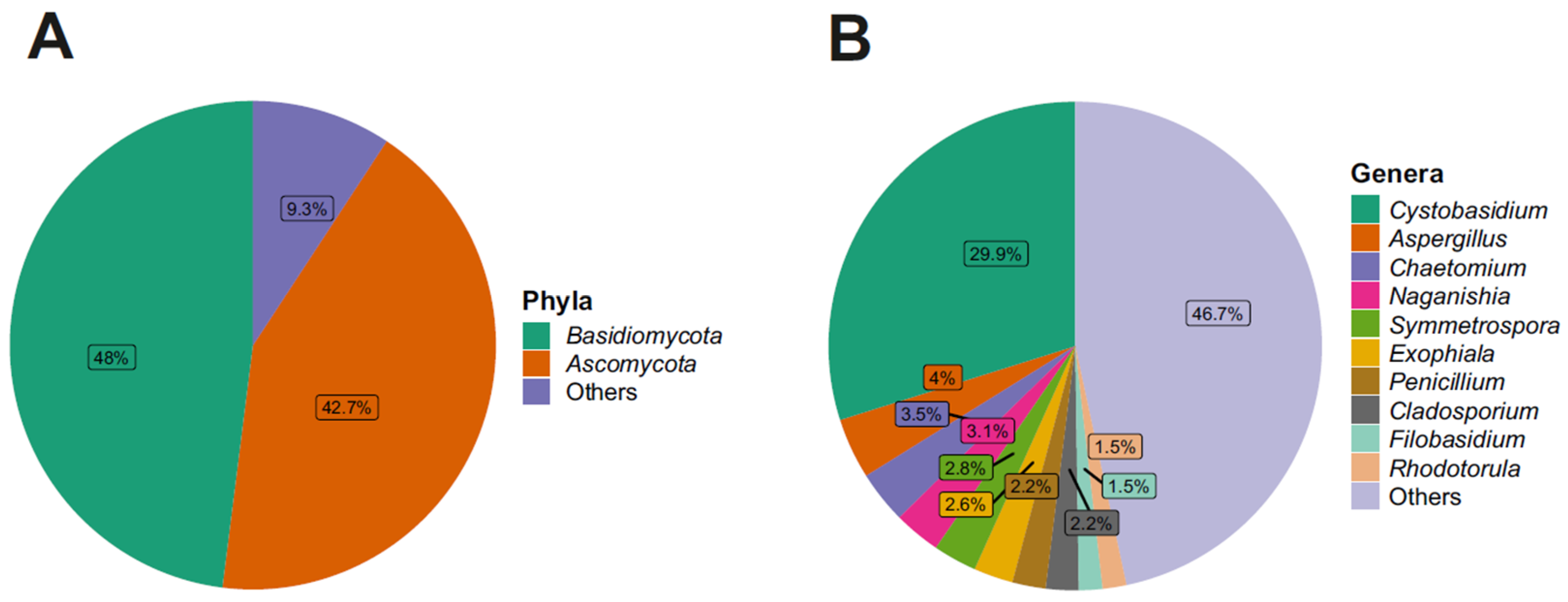
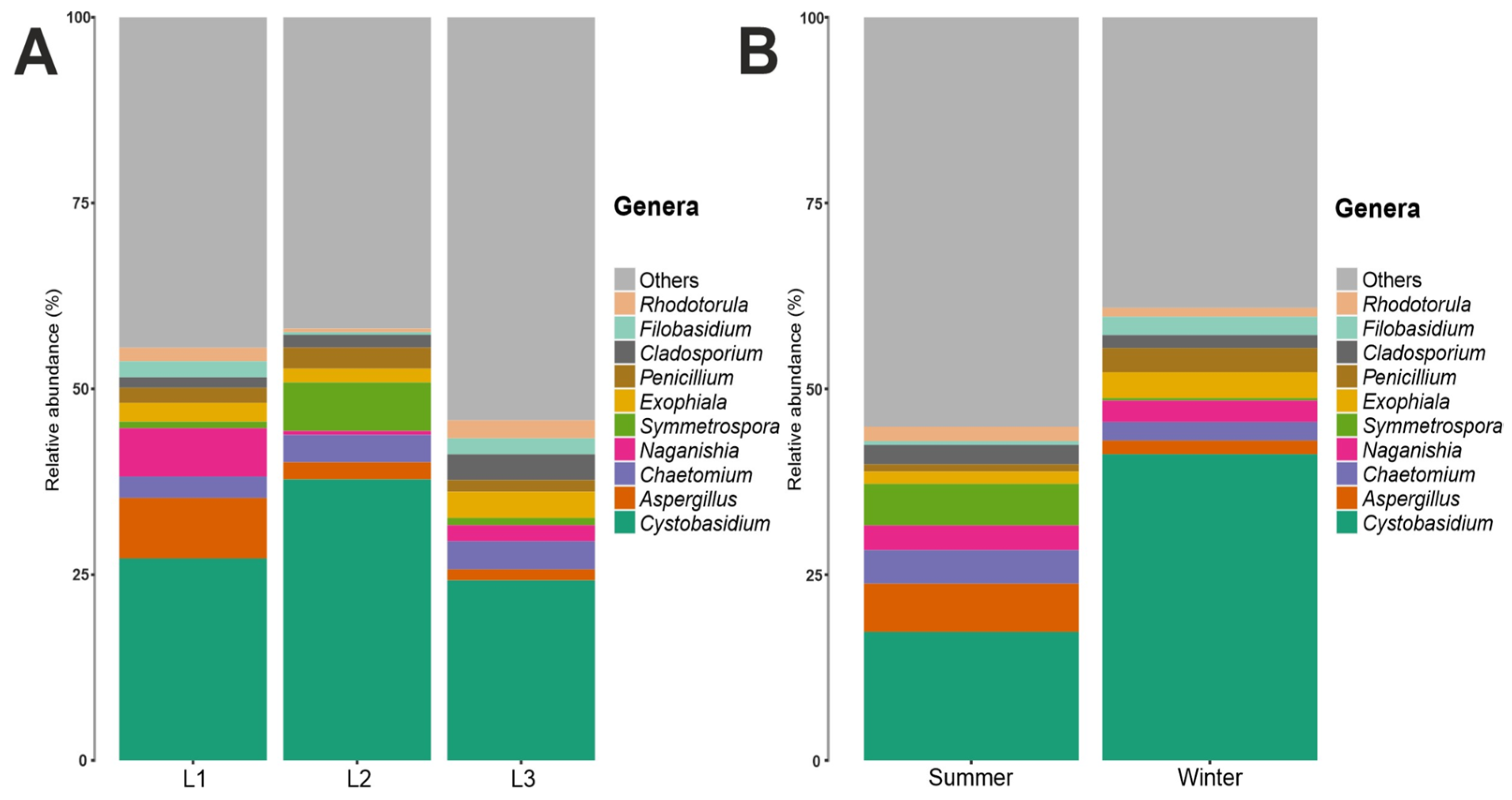
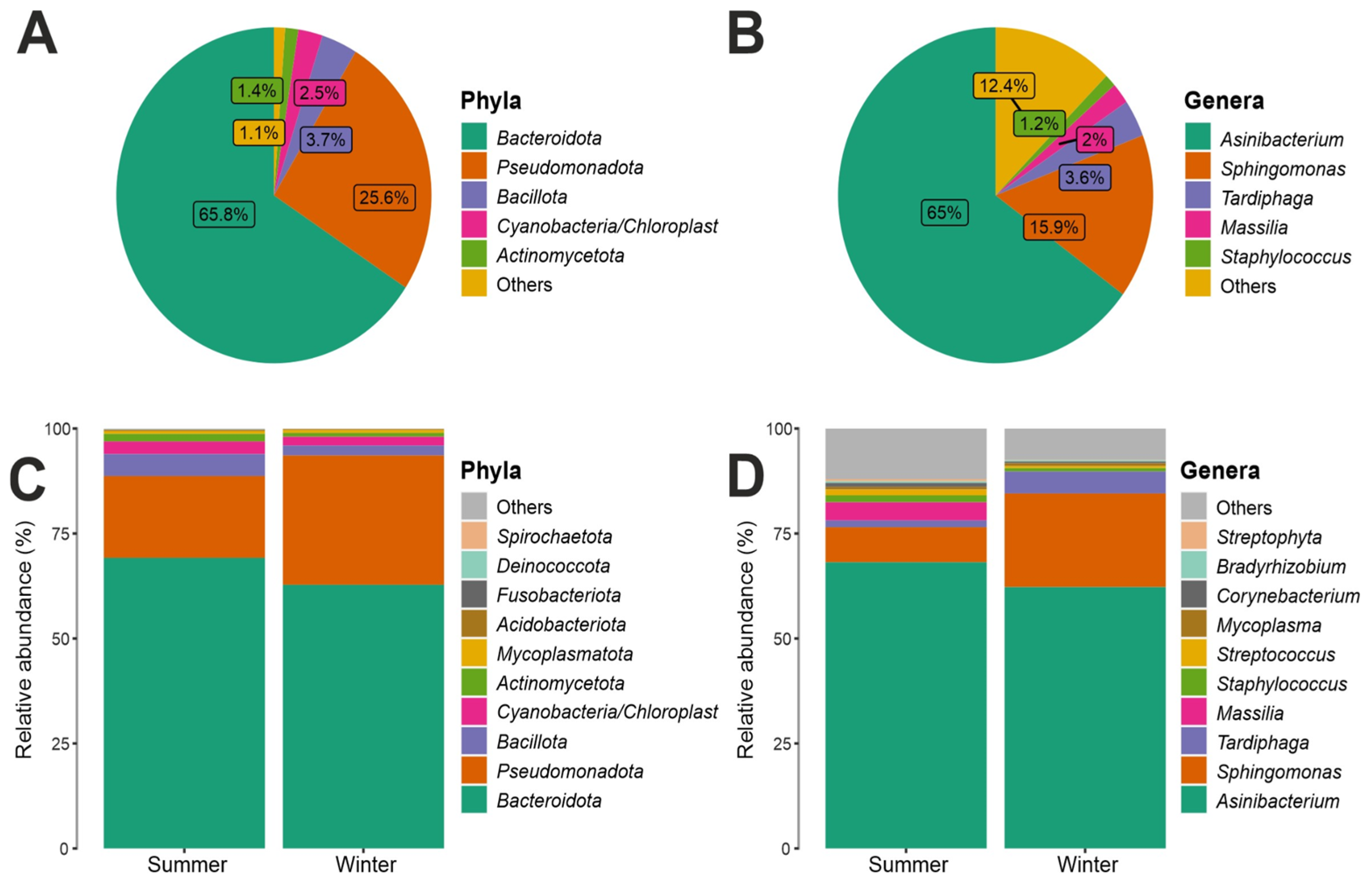
3.2. Taxonomy Structure and Composition
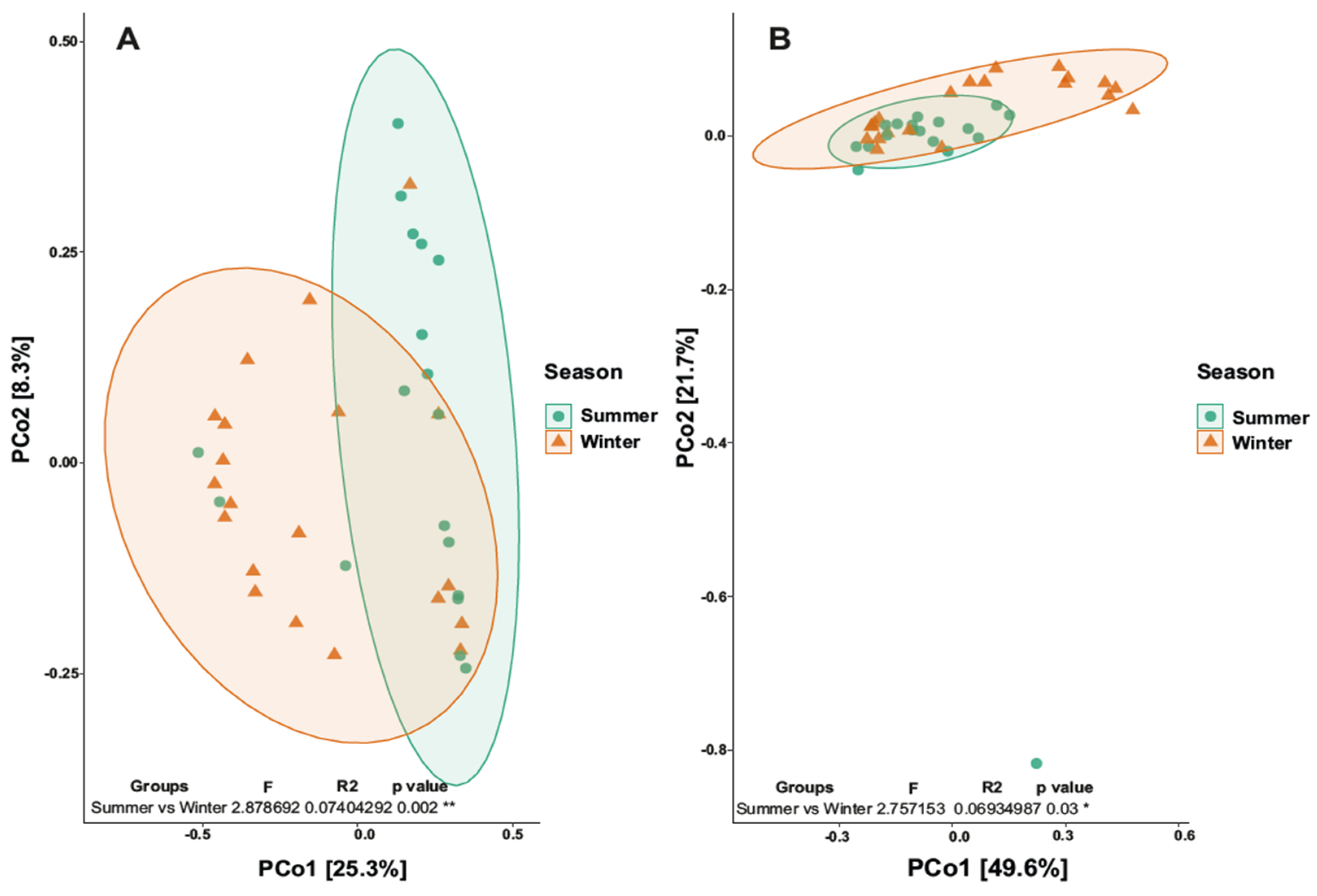
3.3. Biodiversity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearce, D.A.; Alekhina, I.A.; Terauds, A.; Wilmotte, A.; Quesada, A.; Edwards, A.; Dommergue, A.; Sattler, B.; Adams, B.J.; Vincent, W.F.; et al. Aerobiology over Antarctica–A new initiative for atmospheric ecology. Front. Microbiol. 2016, 7, 16. [Google Scholar] [CrossRef]
- Musilova, M.; Wright, G.; Ward, J.M.; Dartnell, L.R. Isolation of Radiation-Resistant Bacteria from Mars Analog Antarctic Dry Valleys by Preselection, and the Correlation between Radiation and Desiccation Resistance. Astrobiology 2015, 15, 1076–1090. [Google Scholar] [CrossRef]
- Romanovskaya, V.A.; Tashirev, A.B.; Shilin, N.; Chernaya, N.A.; Rokitko, P.V.; Levishko, A.S. Resistance of Antarctic microorganisms to UV radiation. Mikrobiolohichnyi Zhurnal 2011, 73, 3–8. [Google Scholar]
- Cassaro, A.; Pacelli, C.; Aureli, L.; Catanzaro, I.; Leo, P.; Onofri, S. Antarctica as a reservoir of planetary analogue environments. Extremophiles 2021, 25, 437–458. [Google Scholar] [CrossRef]
- Miteva, V.I.; Sheridan, P.P.; Brenchley, J.E. Phylogenetic and Physiological Diversity of Microorganisms Isolated from a Deep Greenland Glacier Ice Core. Appl. Environ. Microbiol. 2004, 70, 202–213. [Google Scholar] [CrossRef]
- Napoli, A.; Coleine, C.; Ulrich, N.J.; Moeller, R.; Billi, D.; Selbmann, L. Snow Surface Microbial Diversity at the Detection Limit within the Vicinity of the Concordia Station, Antarctica. Life 2022, 13, 113. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbrück, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Fuhran, J.A.; Field, D.; et al. Defining seasonal marine microbial community dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Taylor, B. Isolation of plant DNA and RNA. Focus 1982, 4, 4–6. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2012, 7, 335–336. [Google Scholar] [CrossRef]
- Smith, D.P.; Peay, K.G. Sequence Depth, Not PCR Replication, Improves Ecological Inference from Next Generation DNA Sequencing. PLoS ONE 2014, 9, e90234. [Google Scholar] [CrossRef]
- Palmer, J.M.; Jusino, M.A.; Banik, M.T.; Lindner, D.L. Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. PeerJ 2018, 6, e4925. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Morris, E.K.; Caruso, T.; Buscot, F.; Fischer, M.; Hancock, C.; Maier, T.S.; Meiners, T.; Müller, C.; Obermaier, E.; Prati, D.; et al. Choosing and using diversity indices: Insights for ecological applications from the German Biodiversity Exploratories. Ecol. Evol. 2014, 4, 3514–3524. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community ecology package (version 2.5-6). Compr. R Arch. Netw. 2019, 3, 33. [Google Scholar]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; The University of Illinois: Urbana, IL, USA; London, UK, 1949; pp. 3–24. [Google Scholar]
- Ludwig, J.A.; Reynolds, J.F.; Quartet, L.; Reynolds, J.F. Statistical Ecology: A Primer in Methods and Computing; John Wiley & Sons: Hoboken, NJ, USA, 1988; Volume 1. [Google Scholar]
- Buzzini, P.; Turk, M.; Perini, L.; Turchetti, B.; Gunde-Cimerman, N. Lieviti in habitat polari e sub-polari. In Lieviti Negli Ecosistemi Naturali: Diversità; Buzzini, P., Lachance, M.A., Yurkov, A., Eds.; Springer: Cham, Swizterland, 2017; pp. 331–365. [Google Scholar]
- Buzzini, P.; Branda, E.; Goretti, M.; Turchetti, B. Psychrophilic yeasts from worldwide glacial habitats: Diversity, adaptation strategies and biotechnological potential. FEMS Microbiol. Ecol. 2012, 82, 217–241. [Google Scholar] [CrossRef]
- Selbmann, L.; Zucconi, L.; Onofri, S.; Cecchini, C.; Isola, D.; Turchetti, B.; Buzzini, P. Taxonomic and phenotypic characterization of yeasts isolated from worldwide cold rock-associated habitats. Fungal Biol. 2014, 118, 61–71. [Google Scholar] [CrossRef]
- Sampaio, J.P.; Agerer, R.; Piepenbring, M.; Blanz, P. Diversity, phylogeny and classification of basidiomycetous yeasts. Front. Basidiomycote Mycol. 2004, 1, 49–80. [Google Scholar]
- Liebkind, D.; Sampaio, J.P.; van Broock, M. Cystobasidiomycetes yeasts from Patagonia (Argentina): Description of Rhodotorula meli sp. nov. from glacial meltwater. Int. J. Syst. Evol. Microbiol. 2010, 60, 2251–2256. [Google Scholar] [CrossRef]
- Turchetti, B.; Goretti, M.; Branda, E.; Diolaiuti, G.; D’Agata, C.; Smiraglia, C.; Onofri, A.; Buzzini, P. Influence of abiotic var-iables on culturable yeast diversity in two distinct Alpine glaciers. FEMS Microbiol. Ecol. 2013, 86, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Vishniac, H.S. Yeast biodiversity in the Antarctic. In Biodiversity and Ecophysiology of Yeasts; Springer: Berlin/Heidelberg, Germany, 2006; pp. 419–440. [Google Scholar]
- Shivaji, S.; Prasad, G.S. Antarctic Yeasts: Biodiversity and Potential Applications. In Yeast Biotechnology: Diversity and Applications; Springer: Dordrecht, The Netherlands, 2009; pp. 3–18. [Google Scholar]
- Tsuji, M.; Tsujimoto, M.; Imura, S. Cystobasidium tubakii and Cystobasidium ongulense, new basidiomycetous yeast species isolated from East Ongul Island, East Antarctica. Mycoscience 2017, 58, 103–110. [Google Scholar] [CrossRef]
- Laich, F.; Vaca, I.; Chávez, R. Rhodotorula portillonensis sp. nov., a basidiomycetous yeast isolated from Antarctic shallow-water marine sediment. Int. J. Syst. Evol. Microbiol. 2013, 63, 3884–3891. [Google Scholar] [CrossRef] [PubMed]
- Turchetti, B.; Selbmann, L.; Gunde-Cimerman, N.; Buzzini, P.; Sampaio, J.P.; Zalar, P. Cystobasidium alpinum sp. nov. and Rhodosporidiobolus oreadorum sp. nov. from European Cold Environments and Arctic Region. Life 2018, 8, 9. [Google Scholar] [CrossRef]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse Lifestyles and Strategies of Plant Pathogenesis Encoded in the Genomes of Eighteen Dothideomycetes Fungi. PLoS Pathog. 2012, 8, e1003037. [Google Scholar] [CrossRef] [PubMed]
- Coleine, C.; Zucconi, L.; Onofri, S.; Pombubpa, N.; Stajich, J.E.; Selbmann, L. Sun Exposure Shapes Functional Grouping of Fungi in Cryptoendolithic Antarctic Communities. Life 2018, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.M.; Cannon, P.F.; Minter, D.W.; Stalpers, J.A. Dictionary of the Fungi, 10th ed.; CABIL: Wallingford, UK, 2008; p. 22. [Google Scholar]
- Shivaji, S.; Begum, Z.; Rao, S.S.S.N.; Reddy, P.V.V.V.; Manasa, P.; Sailaja, B.; Prathiba, M.S.; Thamban, M.; Krishnan, K.P.; Singh, S.M.; et al. Antarctic ice core samples: Culturable bacterial diversity. Res. Microbiol. 2013, 164, 70–82. [Google Scholar] [CrossRef]
- Michaud, L.; Caruso, C.; Mangano, S.; Interdonato, F.; Bruni, V.; Lo Giudice, A. Predominance of Flavobacterium, Pseudomonas, and Polaromonas within the prokaryotic community of freshwater shallow lakes in the northern Victoria Land, East Antarctica. FEMS Microbiol. Ecol. 2012, 82, 391–404. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Costello, E.K.; Hamady, M.; Lozupone, C.; Jiang, L.; Schmidt, S.K.; Fierer, N.; Townsend, A.R.; Cleveland, C.C.; Stanish, L.; et al. Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 2011, 13, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Dong, Q.; Shen, W.; Liu, X.; Dou, N.; Xian, L.; Chen, H. Variation in Near-Surface Airborne Bacterial Communities among Five Forest Types. Forests 2020, 11, 561. [Google Scholar] [CrossRef]
- Tang, K.; Huang, Z.; Huang, J.; Maki, T.; Zhang, S.; Shimizu, A.; Ma, X.; Shi, J.; Bi, J.; Zhang, L. Characterization of atmospheric bio-aerosols along the transport pathway of Asian dust during the Dust-Bioaerosol 2016 Campaign. Atmos. Chem. Phys. 2018, 18, 7131–7148. [Google Scholar] [CrossRef]
- Albanese, D.; Coleine, C.; Rota-Stabelli, O.; Onofri, S.; Tringe, S.G.; Stajich, J.E.; Selbmann, L.; Donati, C. Pre-Cambrian roots of novel Antarctic cryptoendolithic bacterial lineages. Microbiome 2021, 9, 63. [Google Scholar] [CrossRef]
- Lee, D.G.; Park, J.M.; Kang, H.; Hong, S.Y.; Lee, K.R.; Chang, H.B.; Trujillo, M.E. Asinibacteriumlactis gen. nov., sp. nov., a member of the family Chitinophagaceae, isolated from donkey (Equusasinus) milk. Int. J. Syst. Evol. 2013, 63, 3180–3185. [Google Scholar] [CrossRef]
- Brzoska, R.M.; Edelmann, R.E.; Bollmann, A. Physiological and Genomic Characterization of two novel bacteroi-dota strains Asinibacterium spp. OR43 and OR53. Bacteria 2022, 1, 33–47. [Google Scholar] [CrossRef]
- Magalhães, R.; Guerreiro, I.; Santos, R.A.; Coutinho, F.; Couto, A.; Serra, C.; Olsen, R.E.; Peres, H.; Oliva-Teles, A. Oxidative status and intestinal health of gilthead sea bream (Sparus aurata) juveniles fed diets with different ARA/EPA/DHA ratios. Sci. Rep. 2020, 10, Ulrich1-13. [Google Scholar] [CrossRef]
- Fay, M.; Salazar, J.K.; Ramachandran, P.; Stewart, D. Microbiomes of commercially-available pine nuts and sesame seeds. PLoS ONE 2021, 16, e0252605. [Google Scholar] [CrossRef]
- Antony, R.; Sanyal, A.; Kapse, N.; Dhakephalkar, P.K.; Thamban, M.; Nair, S. Microbial communities associated with Antarctic snow pack and their biogeochemical implications. Microbiol. Res. 2016, 192, 192–202. [Google Scholar] [CrossRef]
- Chong, C.-W.; Pearce, D.A.; Convey, P. Emerging spatial patterns in Antarctic prokaryotes. Front. Microbiol. 2015, 6, 1058. [Google Scholar] [CrossRef]
- Van Houdt, R.; De Boever, P.; Coninx, I.; Le Calvez, C.; Dicasillati, R.; Mahillon, J.; Mergeay, M.; Leys, N. Evaluation of the Airborne Bacterial Population in the Periodically Confined Antarctic Base Concordia. Microb. Ecol. 2009, 57, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Els, N.; Larose, C.; Baumann-Stanzer, K.; Tignat-Perrier, R.; Keuschnig, C.; Vogel, T.M.; Sattler, B. Microbial com-position in seasonal time series of free tropospheric air and precipitation reveals community separation. Aerobiologia 2019, 35, 671–701. [Google Scholar] [CrossRef]
- McNeill, V.F.; Grannas, A.M.; Abbatt, J.P.; Ammann, M.; Ariya, P.; Bartels-Rausch, T.; Domine, F.; Donaldson, D.J.; Guzman, M.I.; Heger, D.; et al. Organics in environmental ices: Sources, chemistry, and impacts. Atmos. Chem. Phys. 2012, 12, 9653–9678. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoppiello, G.A.; Coleine, C.; Moeller, R.; Ripa, C.; Billi, D.; Selbmann, L. Seasonality Is the Main Determinant of Microbial Diversity Associated to Snow/Ice around Concordia Station on the Antarctic Polar Plateau. Biology 2023, 12, 1193. https://doi.org/10.3390/biology12091193
Stoppiello GA, Coleine C, Moeller R, Ripa C, Billi D, Selbmann L. Seasonality Is the Main Determinant of Microbial Diversity Associated to Snow/Ice around Concordia Station on the Antarctic Polar Plateau. Biology. 2023; 12(9):1193. https://doi.org/10.3390/biology12091193
Chicago/Turabian StyleStoppiello, Gerardo A., Claudia Coleine, Ralf Moeller, Caterina Ripa, Daniela Billi, and Laura Selbmann. 2023. "Seasonality Is the Main Determinant of Microbial Diversity Associated to Snow/Ice around Concordia Station on the Antarctic Polar Plateau" Biology 12, no. 9: 1193. https://doi.org/10.3390/biology12091193