
Interaction between Autophagy and Senescence in Pancreatic Beta Cells
Abstract
:Simple Summary
Abstract
1. Introduction
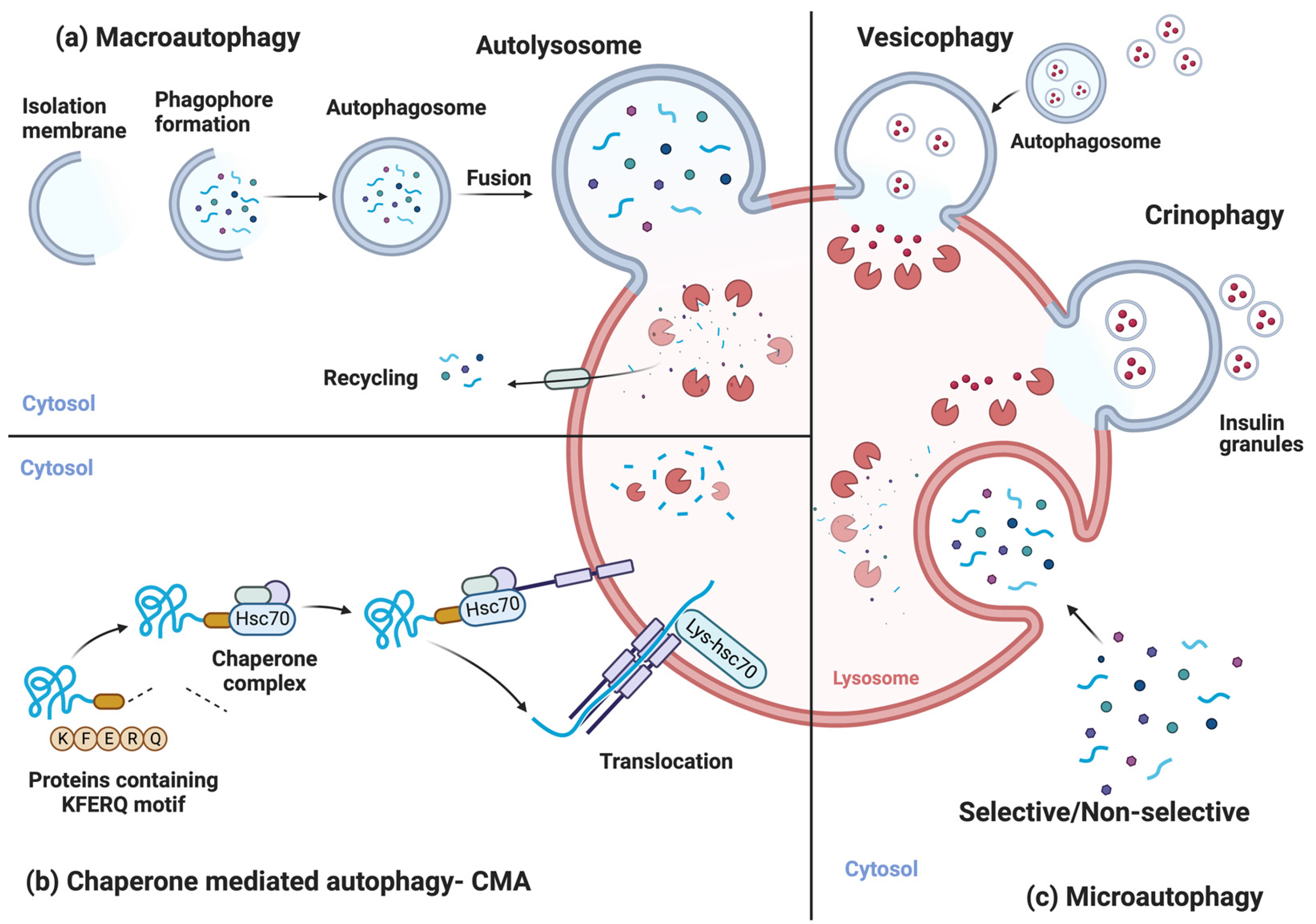
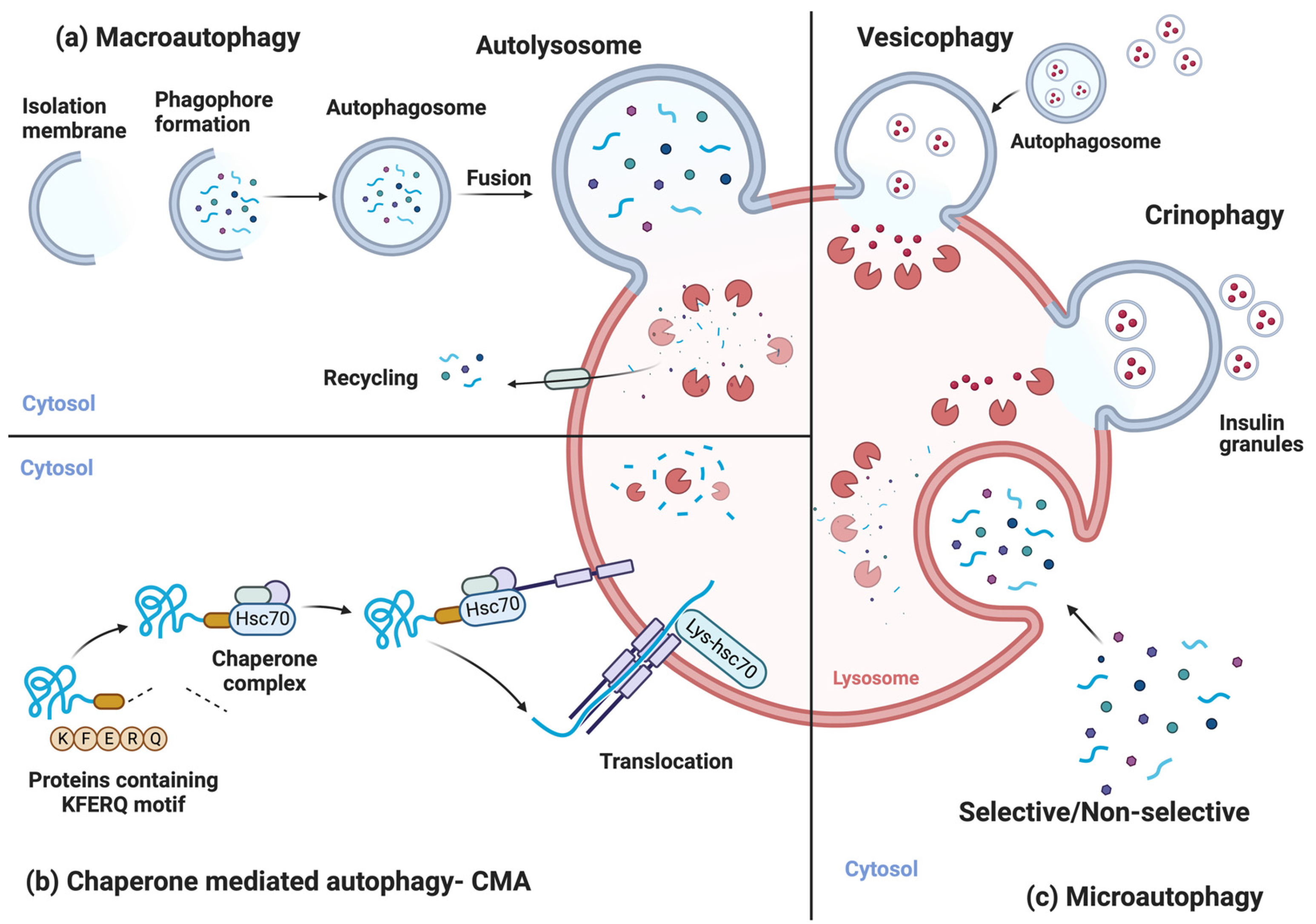
2. Autophagic Processes
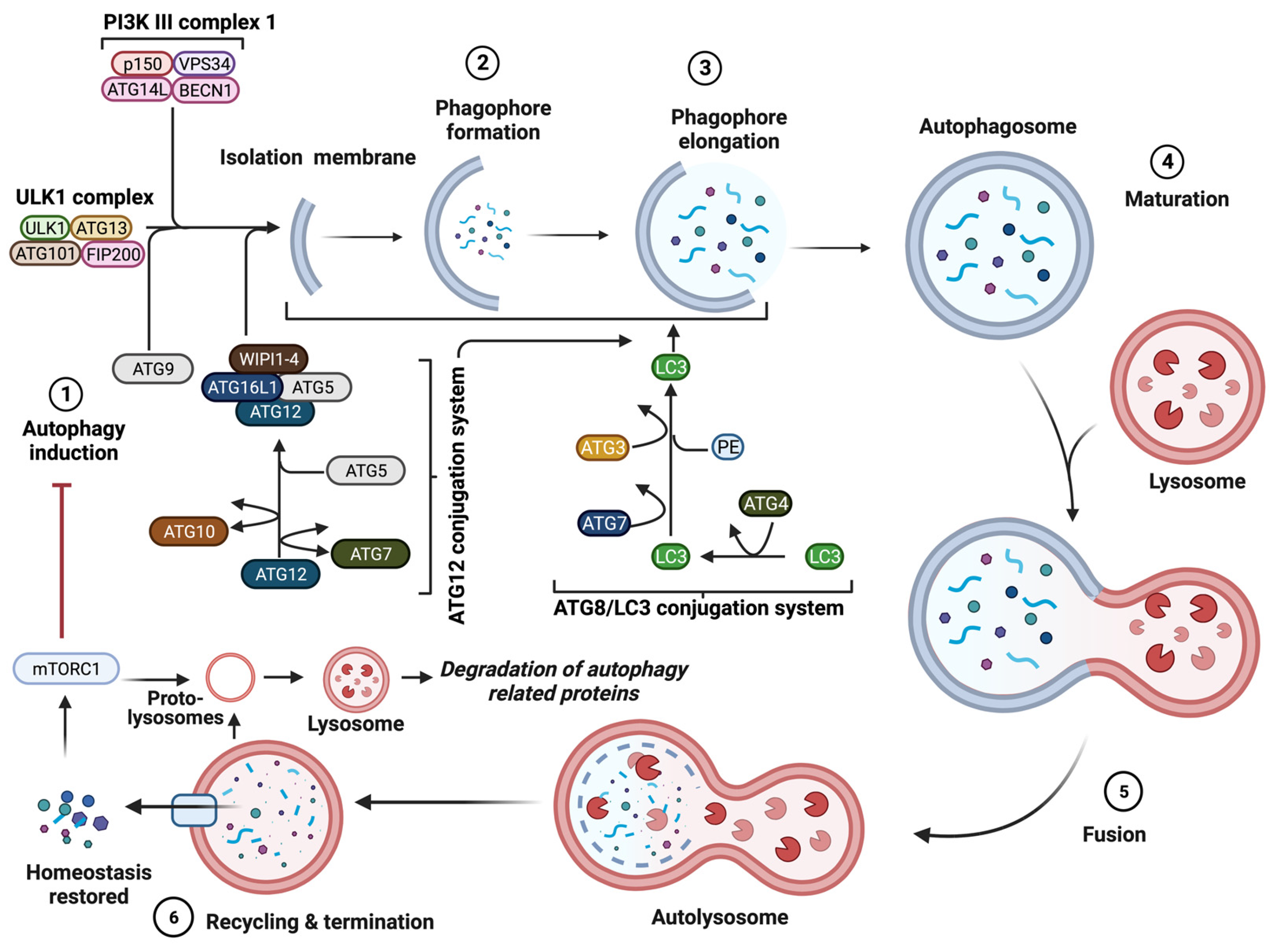
3. Molecular Mechanism of Autophagy
4. Autophagy in β-Cell and Diabetes
5. Cellular Senescence
6. Cellular Senescence in β-Cells and Diabetes
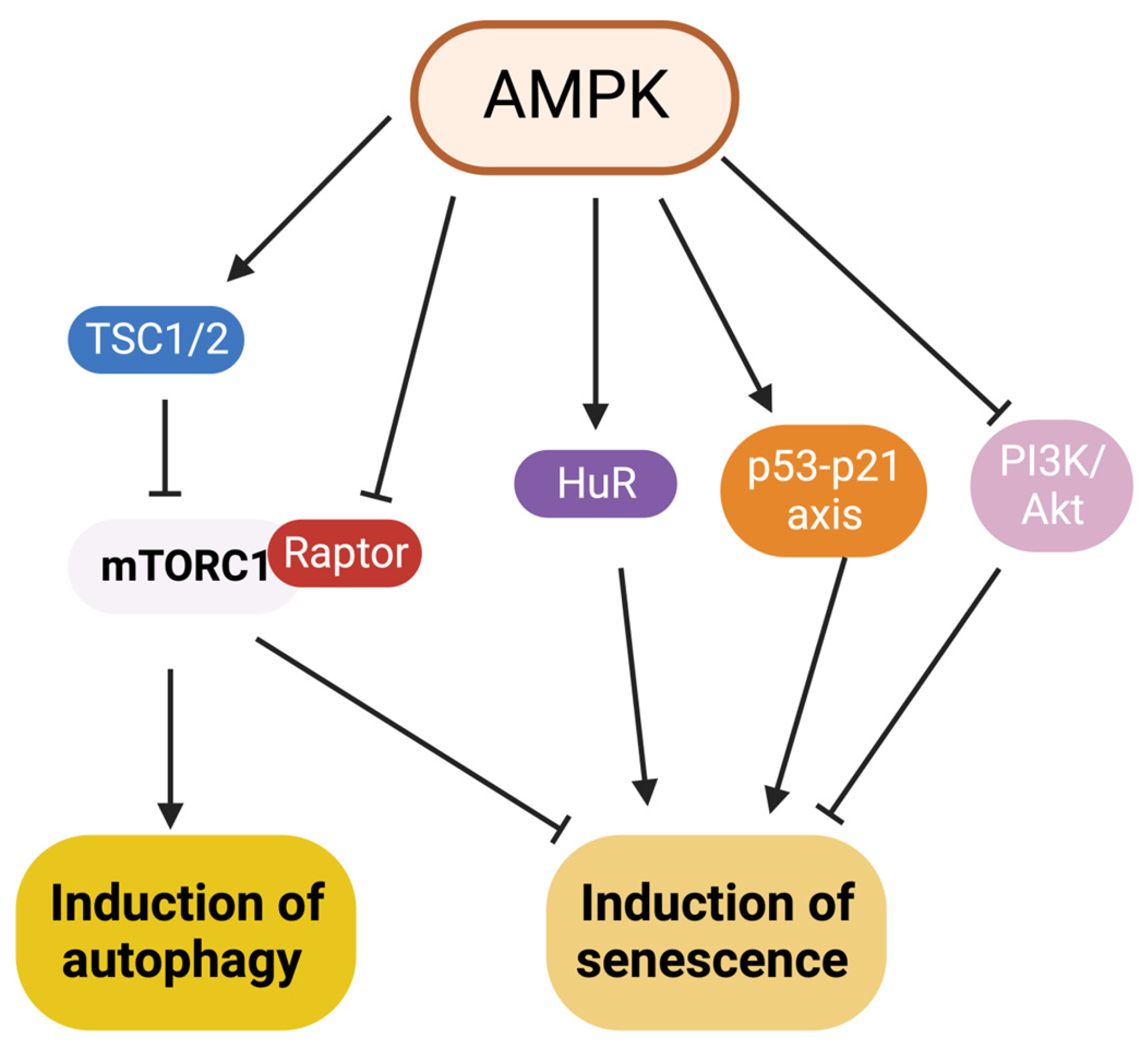
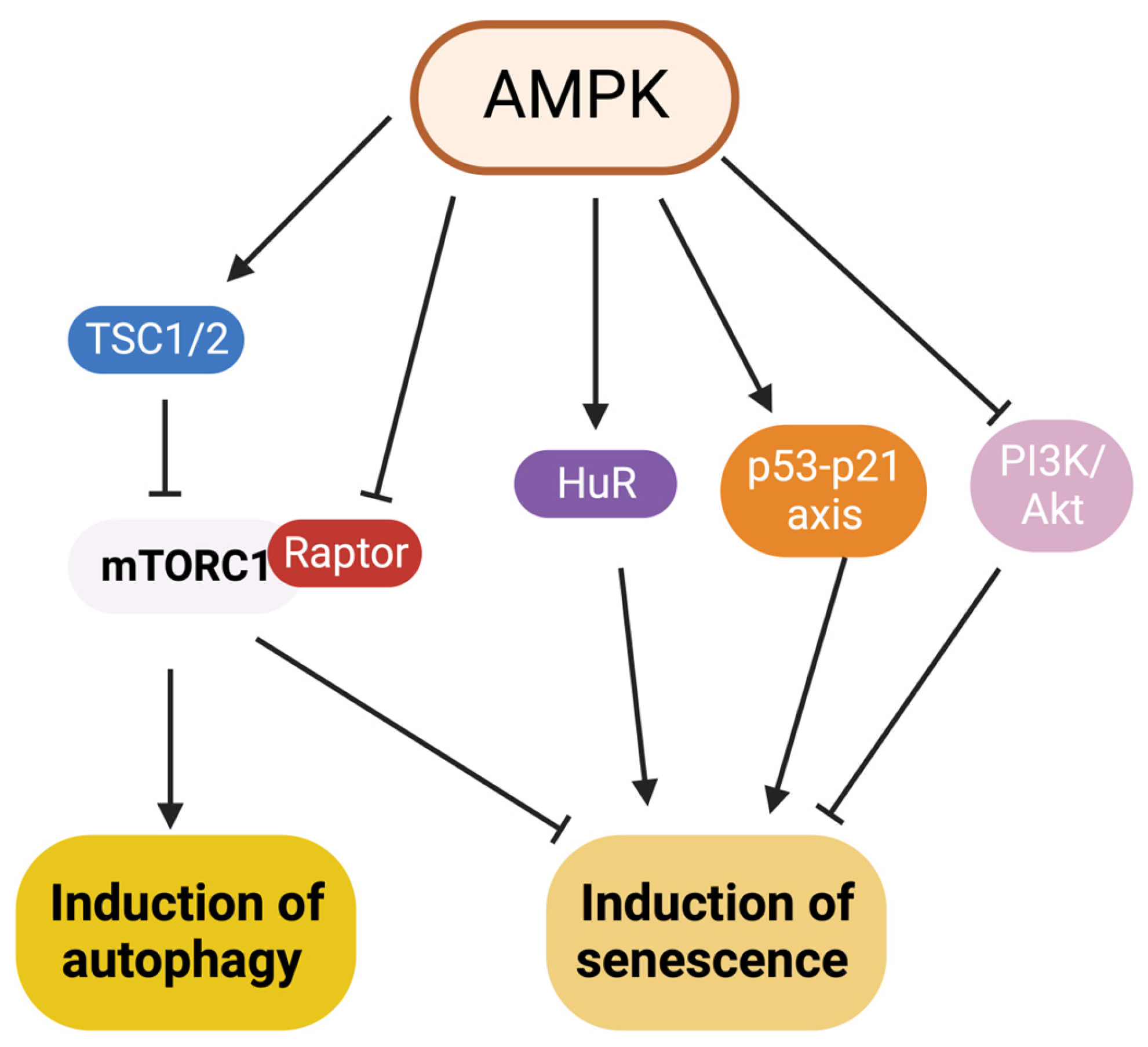
7. Interaction between Autophagy and Senescence

{kind=link}
{kind=link}
{kind=link}
| Autophagy Process | Type of Senescence | Cell Type | Ref. |
|---|---|---|---|
| Macroautophagy | Toxic cellular waste accumulation, resulting in entry into senescence | Primary murine muscle satellite cell | [99] |
| Macroautophagy | Premature senescence | Primary human diploid fibroblasts | [100] |
| Macroautophagy | Oxidative stress-induced senescence | NIH3T3 and MRC-5 cell lines | [102] |
| Macroautophagy | Oxidative stress-induced senescence | NIH3T, MRC-5 and HUVEC cell lines | [103] |
| Mitophagy | Mitochondrial dysfunction-associated senescence (MiDAS) | IMR90, BJ cell lines and Inguinal adipose tissue | [104] |
| Mitophagy | Oxidative stress-induced senescence | Human diploid and ataxia telangiectasia fibroblasts | [105] |
| p62-mediated selective macroautophagy | DNA damage response- induced senescence | IMR90 and BJ cell lines | [107] |
| Macroautophagy | Oxidative stress-induced senescence | SH-SY5Y cell line | [108] |
| Macroautophagy | Oncogene-induced senescence | MEF cell line | [109] |
| Autophagy Process | Type of Senescence | Cell Type | Ref. |
|---|---|---|---|
| Macroautophagy | Oncogene-induced senescence | IMR90, BJ cell lines | [110] |
| mTOR-autophagy spatial coupling compartment | Oncogene-induced senescence | IMR90 cell line | [111] |
| Macroautophagy | Genetically inhibited senescence | hTERT-BJ1 and MDA-MB-231 cell lines | [112] |
| Macroautophagy | Oncogene-induced senescence | Melanoma cells and primary melanocytes | [113] |
| Macroautophagy | Oncogene-induced senescence | IMR90, BJ cell lines | [114] |
| Macroautophagy | Chemotherapy-induced senescence | MCF-7 and HTC-116 cell lines | [115] |
| Macroautophagy | Replicative senescence | MRC-5, WI-38 and p53-null immortalized fibroblast cell lines | [116,117] |
| Macroautophagy | Radiation-induced senescence | AMC-HN-9, U-87, U251, A549, HT-29, MDA-MB-231 cell lines | [118] |
| Macroautophagy | Radiation-induced senescence | MDA-MB-231 and PTTG1-knockdown MDA-MB-231-2A cell lines | [119] |
8. Are Autophagy and Senescence a Potential Therapeutical Target in Diabetes?
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, A.; Flores, R.R.; Robbins, P.D.; Niedernhofer, L.J. Role of Cellular Senescence in Type II Diabetes. Endocrinology 2021, 162, bqab136. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C. Functional changes in beta cells during ageing and senescence. Diabetologia 2020, 63, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Montegut, L.; Abdellatif, M.; Motino, O.; Madeo, F.; Martins, I.; Quesada, V.; Lopez-Otin, C.; Kroemer, G. Acyl coenzyme A binding protein (ACBP): An aging- and disease-relevant “autophagy checkpoint”. Aging Cell 2023, e13910. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060.e10. [Google Scholar] [CrossRef]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Yoshimori, T. Autophagosome biogenesis and human health. Cell Discov. 2020, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Zhang, S.; Mizushima, N. Autophagy genes in biology and disease. Nat. Rev. Genet. 2023, 24, 382–400. [Google Scholar] [CrossRef] [PubMed]
- Weckman, A.; Di Ieva, A.; Rotondo, F.; Syro, L.V.; Ortiz, L.D.; Kovacs, K.; Cusimano, M.D. Autophagy in the endocrine glands. J. Mol. Endocrinol. 2014, 52, R151–R163. [Google Scholar] [CrossRef] [PubMed]
- Kuramoto, K.; He, C. The BECN1-BCL2 complex regulates insulin secretion and storage in mice. Autophagy 2018, 14, 2026–2028. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Makino, S.; Kagohashi, Y.; Sasaki, M.; Ohsumi, Y. A method for the isolation and characterization of autophagic bodies from yeast provides a key tool to investigate cargos of autophagy. J. Biol. Chem. 2022, 298, 102641. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 2015, 520, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Yim, W.W.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Chen, Z.F.; Li, Y.B.; Han, J.Y.; Yin, J.J.; Wang, Y.; Zhu, L.B.; Xie, G.Y. Liraglutide prevents high glucose level induced insulinoma cells apoptosis by targeting autophagy. Chin. Med. J. 2013, 126, 937–941. [Google Scholar]
- Abe, H.; Uchida, T.; Hara, A.; Mizukami, H.; Komiya, K.; Koike, M.; Shigihara, N.; Toyofuku, Y.; Ogihara, T.; Uchiyama, Y.; et al. Exendin-4 improves beta-cell function in autophagy-deficient beta-cells. Endocrinology 2013, 154, 4512–4524. [Google Scholar] [CrossRef] [PubMed]
- Marsh, B.J.; Soden, C.; Alarcon, C.; Wicksteed, B.L.; Yaekura, K.; Costin, A.J.; Morgan, G.P.; Rhodes, C.J. Regulated autophagy controls hormone content in secretory-deficient pancreatic endocrine beta-cells. Mol. Endocrinol. 2007, 21, 2255–2269. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.M.; Lim, H.; Hur, K.Y.; Quan, W.; Lee, H.Y.; Cheon, H.; Ryu, D.; Koo, S.H.; Kim, H.L.; Kim, J.; et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat. Commun. 2014, 5, 4934. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Kuramoto, K.; Wang, N.; Situ, X.; Priyadarshini, M.; Zhang, W.; Cordoba-Chacon, J.; Layden, B.T.; He, C. Autophagy Differentially Regulates Insulin Production and Insulin Sensitivity. Cell Rep. 2018, 23, 3286–3299. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef]
- Corsa, C.A.S.; Pearson, G.L.; Renberg, A.; Askar, M.M.; Vozheiko, T.; MacDougald, O.A.; Soleimanpour, S.A. The E3 ubiquitin ligase parkin is dispensable for metabolic homeostasis in murine pancreatic beta cells and adipocytes. J. Biol. Chem. 2019, 294, 7296–7307. [Google Scholar] [CrossRef]
- Houck, S.A.; Ren, H.Y.; Madden, V.J.; Bonner, J.N.; Conlin, M.P.; Janovick, J.A.; Conn, P.M.; Cyr, D.M. Quality control autophagy degrades soluble ERAD-resistant conformers of the misfolded membrane protein GnRHR. Mol. Cell 2014, 54, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Vander Mierde, D.; Song, B.; Flamez, D.; Creemers, J.W.; Tsukamoto, K.; Ribick, M.; Schuit, F.C.; Kaufman, R.J. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat. Med. 2005, 11, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Kaniuk, N.A.; Kiraly, M.; Bates, H.; Vranic, M.; Volchuk, A.; Brumell, J.H. Ubiquitinated-protein aggregates form in pancreatic beta-cells during diabetes-induced oxidative stress and are regulated by autophagy. Diabetes 2007, 56, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Ariav, Y.; Tirosh, B.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 2013, 62, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.C.; Kim, K.H.; Kim, G.H.; Kim, S.W.; Kim, H.L.; Lee, M.K.; et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.E.; Lee, S.M.; Lee, Y.J.; Li, L.J.; Lee, S.J.; Lee, J.H.; Kim, Y.; Jun, H.S.; Lee, K.W.; Kang, Y. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology 2009, 150, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Yang, B.; Olson, L.K.; Greenstein, A.; Baek, S.H.; Claycombe, K.J.; Goudreau, J.L.; Yu, S.W.; Kim, E.K. Activation of autophagy through modulation of 5’-AMP-activated protein kinase protects pancreatic beta-cells from high glucose. Biochem. J. 2010, 425, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef]
- Fujimoto, K.; Hanson, P.T.; Tran, H.; Ford, E.L.; Han, Z.; Johnson, J.D.; Schmidt, R.E.; Green, K.G.; Wice, B.M.; Polonsky, K.S. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J. Biol. Chem. 2009, 284, 27664–27673. [Google Scholar] [CrossRef]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef]
- Bugliani, M.; Mossuto, S.; Grano, F.; Suleiman, M.; Marselli, L.; Boggi, U.; De Simone, P.; Eizirik, D.L.; Cnop, M.; Marchetti, P.; et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front. Endocrinol. 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Webley, K.; Bond, J.A.; Jones, C.J.; Blaydes, J.P.; Craig, A.; Hupp, T.; Wynford-Thomas, D. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol. Cell Biol. 2000, 20, 2803–2808. [Google Scholar] [CrossRef]
- Ge, H.; Ni, S.; Wang, X.; Xu, N.; Liu, Y.; Wang, X.; Wang, L.; Song, D.; Song, Y.; Bai, C. Dexamethasone reduces sensitivity to cisplatin by blunting p53-dependent cellular senescence in non-small cell lung cancer. PLoS ONE 2012, 7, e51821. [Google Scholar] [CrossRef] [PubMed]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef]
- Sone, H.; Kagawa, Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia 2005, 48, 58–67. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Krtolica, A.; Beausejour, C.M.; Parrinello, S.; Hodgson, J.G.; Chin, K.; Desprez, P.Y.; Campisi, J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE 2010, 5, e9188. [Google Scholar] [CrossRef]
- Chen, W.; Kang, J.; Xia, J.; Li, Y.; Yang, B.; Chen, B.; Sun, W.; Song, X.; Xiang, W.; Wang, X.; et al. p53-related apoptosis resistance and tumor suppression activity in UVB-induced premature senescent human skin fibroblasts. Int. J. Mol. Med. 2008, 21, 645–653. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Ness, K.K.; Armstrong, G.T.; Kundu, M.; Wilson, C.L.; Tchkonia, T.; Kirkland, J.L. Frailty in childhood cancer survivors. Cancer 2015, 121, 1540–1547. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef]
- Maskey, R.S.; Kim, M.S.; Baker, D.J.; Childs, B.; Malureanu, L.A.; Jeganathan, K.B.; Machida, Y.; van Deursen, J.M.; Machida, Y.J. Spartan deficiency causes genomic instability and progeroid phenotypes. Nat. Commun. 2014, 5, 5744. [Google Scholar] [CrossRef]
- Zou, H.; Stoppani, E.; Volonte, D.; Galbiati, F. Caveolin-1, cellular senescence and age-related diseases. Mech. Ageing Dev. 2011, 132, 533–542. [Google Scholar] [CrossRef]
- Druelle, C.; Drullion, C.; Desle, J.; Martin, N.; Saas, L.; Cormenier, J.; Malaquin, N.; Huot, L.; Slomianny, C.; Bouali, F.; et al. ATF6alpha regulates morphological changes associated with senescence in human fibroblasts. Oncotarget 2016, 7, 67699–67715. [Google Scholar] [CrossRef]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Titus, S.; Li, F.; Stobezki, R.; Akula, K.; Unsal, E.; Jeong, K.; Dickler, M.; Robson, M.; Moy, F.; Goswami, S.; et al. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci. Transl. Med. 2013, 5, 172ra121. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.K.; Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 2012, 3, 263. [Google Scholar] [CrossRef] [PubMed]
- Starr, M.E.; Saito, M.; Evers, B.M.; Saito, H. Age-Associated Increase in Cytokine Production During Systemic Inflammation-II: The Role of IL-1beta in Age-Dependent IL-6 Upregulation in Adipose Tissue. J. Gerontol. Biol. A Sci. Med. Sci. 2015, 70, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.; Langen, R.; Gurlo, T.; Matveyenko, A.V.; Butler, P.C. beta-Cell failure in type 2 diabetes: A case of asking too much of too few? Diabetes 2013, 62, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Teta, M.; Long, S.Y.; Wartschow, L.M.; Rankin, M.M.; Kushner, J.A. Very slow turnover of beta-cells in aged adult mice. Diabetes 2005, 54, 2557–2567. [Google Scholar] [CrossRef]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human beta-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef]
- Li, N.; Liu, F.; Yang, P.; Xiong, F.; Yu, Q.; Li, J.; Zhou, Z.; Zhang, S.; Wang, C.Y. Aging and stress induced beta cell senescence and its implication in diabetes development. Aging 2019, 11, 9947–9959. [Google Scholar] [CrossRef]
- Guo, N.; Parry, E.M.; Li, L.S.; Kembou, F.; Lauder, N.; Hussain, M.A.; Berggren, P.O.; Armanios, M. Short telomeres compromise beta-cell signaling and survival. PLoS ONE 2011, 6, e17858. [Google Scholar] [CrossRef]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Hallinger, D.; Garcia, E.; Machida, Y.; Chakrabarti, S.; Nadler, J.; Galkina, E.V.; Imai, Y. Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 2014, 57, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; van Haaren, M.; Mruk, M.; Lee, T.B., Jr.; Crawford, C.; Hollister-Lock, J.; Sullivan, B.A.; Johnson, J.W.; Ebrahimi, A.; Dreyfuss, J.M.; et al. β Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab. 2017, 25, 898–910.e895. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Lalani, B.; Kahng, J.; Carapeto, P.; Sanjines, S.; Hela, F.; Abarca, C.; Tsuji, T.; Darcy, J.; Bartke, A.; et al. Decreased IGF1R attenuates senescence and improves function in pancreatic beta-cells. Front. Endocrinol. 2023, 14, 1203534. [Google Scholar] [CrossRef]
- Midha, A.; Pan, H.; Abarca, C.; Andle, J.; Carapeto, P.; Bonner-Weir, S.; Aguayo-Mazzucato, C. Unique Human and Mouse beta-Cell Senescence-Associated Secretory Phenotype (SASP) Reveal Conserved Signaling Pathways and Heterogeneous Factors. Diabetes 2021, 70, 1098–1116. [Google Scholar] [CrossRef]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.J.; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2021, 184, 4838–4839. [Google Scholar] [CrossRef]
- Pinho, A.V.; Rooman, I.; Real, F.X. p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle 2011, 10, 1312–1321. [Google Scholar] [CrossRef]
- Grabliauskaite, K.; Hehl, A.B.; Seleznik, G.M.; Saponara, E.; Schlesinger, K.; Zuellig, R.A.; Dittmann, A.; Bain, M.; Reding, T.; Sonda, S.; et al. p21(WAF1) (/Cip1) limits senescence and acinar-to-ductal metaplasia formation during pancreatitis. J. Pathol. 2015, 235, 502–514. [Google Scholar] [CrossRef]
- Horiguchi, M.; Yoshida, M.; Hirata, K.; Furuyama, K.; Masui, T.; Uemoto, S.; Kawaguchi, Y. Senescence caused by inactivation of the homeodomain transcription factor Pdx1 in adult pancreatic acinar cells in mice. FEBS Lett. 2019, 593, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lahmy, R.; Riha, C.; Yang, C.; Jakubison, B.L.; van Niekerk, J.; Staub, C.; Wu, Y.; Gates, K.; Dong, D.S.; et al. The basic helix-loop-helix transcription factor E47 reprograms human pancreatic cancer cells to a quiescent acinar state with reduced tumorigenic potential. Pancreas 2015, 44, 718–727. [Google Scholar] [CrossRef]
- Tourlakis, M.E.; Zhang, S.; Ball, H.L.; Gandhi, R.; Liu, H.; Zhong, J.; Yuan, J.S.; Guidos, C.J.; Durie, P.R.; Rommens, J.M. In Vivo Senescence in the Sbds-Deficient Murine Pancreas: Cell-Type Specific Consequences of Translation Insufficiency. PLoS Genet. 2015, 11, e1005288. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Gukovsky, I. Autophagy and pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G993–G1003. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Y.; Vanhoutte, P.M. SIRT1 and AMPK in regulating mammalian senescence: A critical review and a working model. FEBS Lett. 2011, 585, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Elledge, S.J. How autophagy both activates and inhibits cellular senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367. [Google Scholar] [CrossRef]
- Takenaka, Y.; Inoue, I.; Nakano, T.; Ikeda, M.; Kakinuma, Y. Prolonged disturbance of proteostasis induces cellular senescence via temporal mitochondrial dysfunction and subsequent mitochondrial accumulation in human fibroblasts. FEBS J. 2022, 289, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Nopparat, C.; Sinjanakhom, P.; Govitrapong, P. Melatonin reverses H2O2-induced senescence in SH-SY5Y cells by enhancing autophagy via sirtuin 1 deacetylation of the RelA/p65 subunit of NF-kappaB. J. Pineal Res. 2017, 63, e12407. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.D.; Lapi, E.; Sullivan, A.; Jia, W.; He, Y.W.; Ratnayaka, I.; Zhong, S.; Goldin, R.D.; Goemans, C.G.; et al. Autophagic activity dictates the cellular response to oncogenic RAS. Proc. Natl. Acad. Sci. USA 2012, 109, 13325–13330. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes. Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Ando, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef]
- Liu, H.; He, Z.; Simon, H.U. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 2014, 10, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The autophagy-senescence connection in chemotherapy: Must tumor cells (self) eat before they sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, I.; Fujita, K.; Jenkins, L.M.; Hiyoshi, Y.; Mondal, A.M.; Vojtesek, B.; Lane, D.P.; Appella, E.; Harris, C.C. Autophagic degradation of the inhibitory p53 isoform Delta133p53alpha as a regulatory mechanism for p53-mediated senescence. Nat. Commun. 2014, 5, 4706. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Mondal, A.M.; Horikawa, I.; Nguyen, G.H.; Kumamoto, K.; Sohn, J.J.; Bowman, E.D.; Mathe, E.A.; Schetter, A.J.; Pine, S.R.; et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 2009, 11, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yang, P.M.; Chuah, Q.Y.; Lee, Y.J.; Hsieh, Y.F.; Peng, C.W.; Chiu, S.J. Autophagy promotes radiation-induced senescence but inhibits bystander effects in human breast cancer cells. Autophagy 2014, 10, 1212–1228. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Mohsen, S.; Sobash, P.T.; Algwaiz, G.F.; Nasef, N.; Al-Zeidaneen, S.A.; Karim, N.A. Autophagy Agents in Clinical Trials for Cancer Therapy: A Brief Review. Curr. Oncol. 2022, 29, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Jurk, D.; Khosla, S.; Kirkland, J.L.; LeBrasseur, N.K.; Miller, J.D.; Passos, J.F.; Pignolo, R.J.; Tchkonia, T.; Niedernhofer, L.J. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 779–803. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, J.J.; Yang, L.J.; Wei, L.X.; Zou, D.J. Rosiglitazone protects against palmitate-induced pancreatic beta-cell death by activation of autophagy via 5’-AMP-activated protein kinase modulation. Endocrine 2013, 44, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Huang, W.; Wang, J.; Xu, Z.; He, J.; Lin, X.; Zhou, Z.; Zhang, J. Metformin plays a dual role in MIN6 pancreatic beta cell function through AMPK-dependent autophagy. Int. J. Biol. Sci. 2014, 10, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Riboulet-Chavey, A.; Diraison, F.; Siew, L.K.; Wong, F.S.; Rutter, G.A. Inhibition of AMP-activated protein kinase protects pancreatic beta-cells from cytokine-mediated apoptosis and CD8+ T-cell-induced cytotoxicity. Diabetes 2008, 57, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brule, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010, 11, 390–401. [Google Scholar] [CrossRef]
- Fraenkel, M.; Ketzinel-Gilad, M.; Ariav, Y.; Pappo, O.; Karaca, M.; Castel, J.; Berthault, M.F.; Magnan, C.; Cerasi, E.; Kaiser, N.; et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008, 57, 945–957. [Google Scholar] [CrossRef]
- Xie, Q.; Lin, Q.; Li, D.; Chen, J. Imatinib induces autophagy via upregulating XIAP in GIST882 cells. Biochem. Biophys. Res. Commun. 2017, 488, 584–589. [Google Scholar] [CrossRef]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J. Clin. Investig. 2014, 124, 3311–3324. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Cadoo, K.A.; Gucalp, A.; Traina, T.A. Palbociclib: An evidence-based review of its potential in the treatment of breast cancer. Breast Cancer 2014, 6, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ablain, J.; Rice, K.; Soilihi, H.; de Reynies, A.; Minucci, S.; de The, H. Activation of a promyelocytic leukemia-tumor protein 53 axis underlies acute promyelocytic leukemia cure. Nat. Med. 2014, 20, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Kanigur Sultuybek, G.; Soydas, T.; Yenmis, G. NF-kappaB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clin. Exp. Pharmacol. Physiol. 2019, 46, 413–422. [Google Scholar] [CrossRef]
- Mun, G.I.; Boo, Y.C. Identification of CD44 as a senescence-induced cell adhesion gene responsible for the enhanced monocyte recruitment to senescent endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2102–H2111. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Bahour, N.; Bleichmar, L.; Abarca, C.; Wilmann, E.; Sanjines, S.; Aguayo-Mazzucato, C. Clearance of p16Ink4a-positive cells in a mouse transgenic model does not change beta-cell mass and has limited effects on their proliferative capacity. Aging 2023, 15, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hela, F.; Aguayo-Mazzucato, C. Interaction between Autophagy and Senescence in Pancreatic Beta Cells. Biology 2023, 12, 1205. https://doi.org/10.3390/biology12091205
Hela F, Aguayo-Mazzucato C. Interaction between Autophagy and Senescence in Pancreatic Beta Cells. Biology. 2023; 12(9):1205. https://doi.org/10.3390/biology12091205
Chicago/Turabian StyleHela, Francesko, and Cristina Aguayo-Mazzucato. 2023. "Interaction between Autophagy and Senescence in Pancreatic Beta Cells" Biology 12, no. 9: 1205. https://doi.org/10.3390/biology12091205
APA StyleHela, F., & Aguayo-Mazzucato, C. (2023). Interaction between Autophagy and Senescence in Pancreatic Beta Cells. Biology, 12(9), 1205. https://doi.org/10.3390/biology12091205