
Iguratimod Ameliorates the Severity of Secondary Progressive Multiple Sclerosis in Model Mice by Directly Inhibiting IL-6 Production and Th17 Cell Migration via Mitigation of Glial Inflammation
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Generation of PLP/CreERT;Cx47fl/fl Mice
2.3. Tamoxifen Injection
2.4. Induction and Clinical Evaluation of EAE
2.5. IGU Treatment In Vivo
2.6. Tissue Preparations
2.7. Histopathological and Immunohistochemical Analyses
2.8. Quantification of Immunohistochemical Images
2.9. Microglial Circularity Analysis
2.10. Flow Cytometry
2.11. Isolation of Splenocytes
2.12. T Cell Proliferation Assay
2.13. Glial Cell Cultures
2.14. Harvesting of Glial Cell Culture Supernatant
2.15. Multiplexed Fluorescence Immunoassay for Cytokines
2.16. Chemokine Analysis by ELISA
2.17. Migration Assay
2.18. Incubation of CD4+ T Cells with IGU
2.19. Statistical Analysis
3. Results
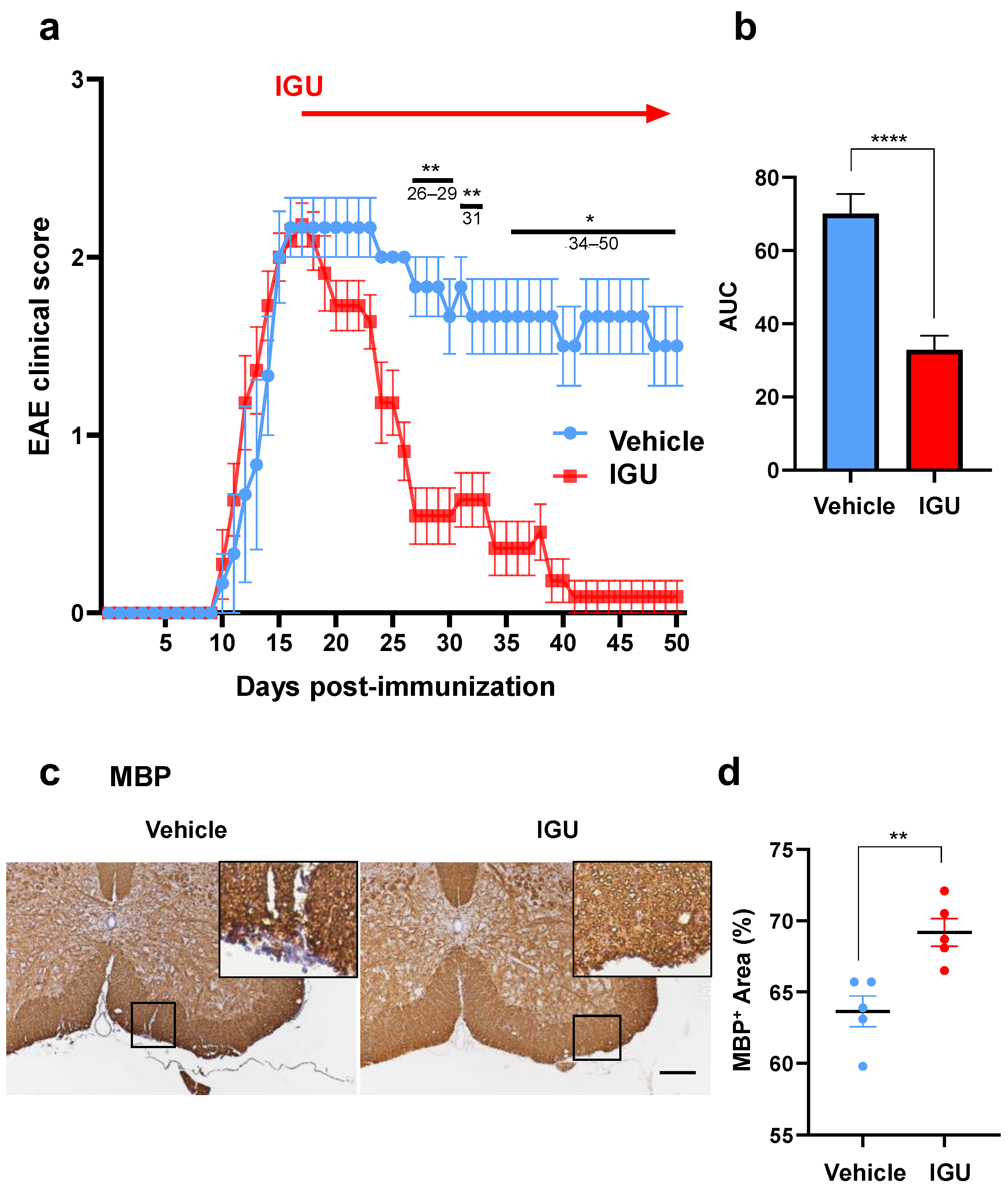
3.1. IGU Ameliorated the Clinical Severity and Demyelination in the Chronic Phase of pEAE
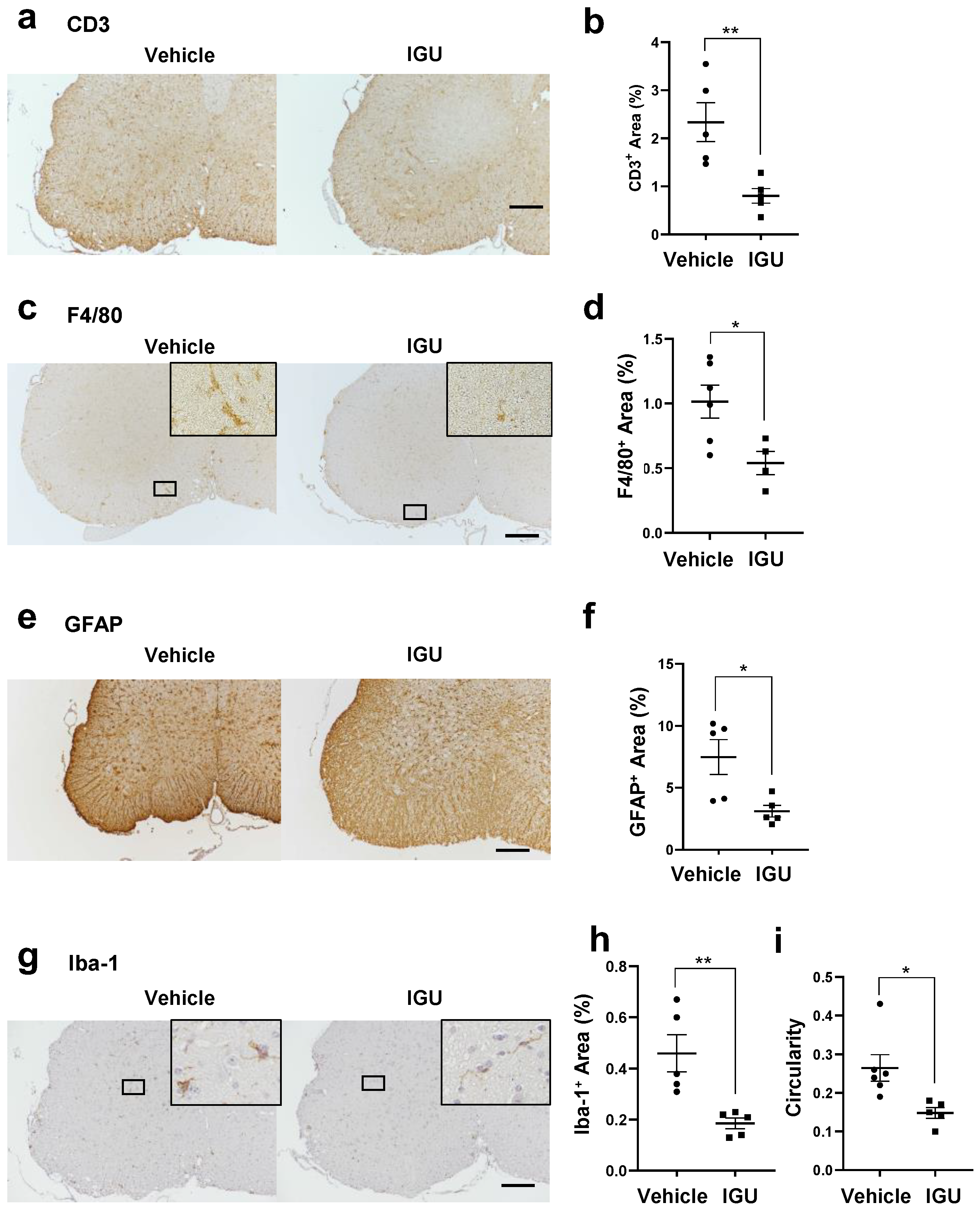
3.2. IGU Inhibited Inflammatory Cell Infiltration of the Lumbar Spinal Cord in the Chronic Phase of pEAE
3.3. IGU Reduced A1 Astroglia and M1-Like Microglia during the Chronic Phase
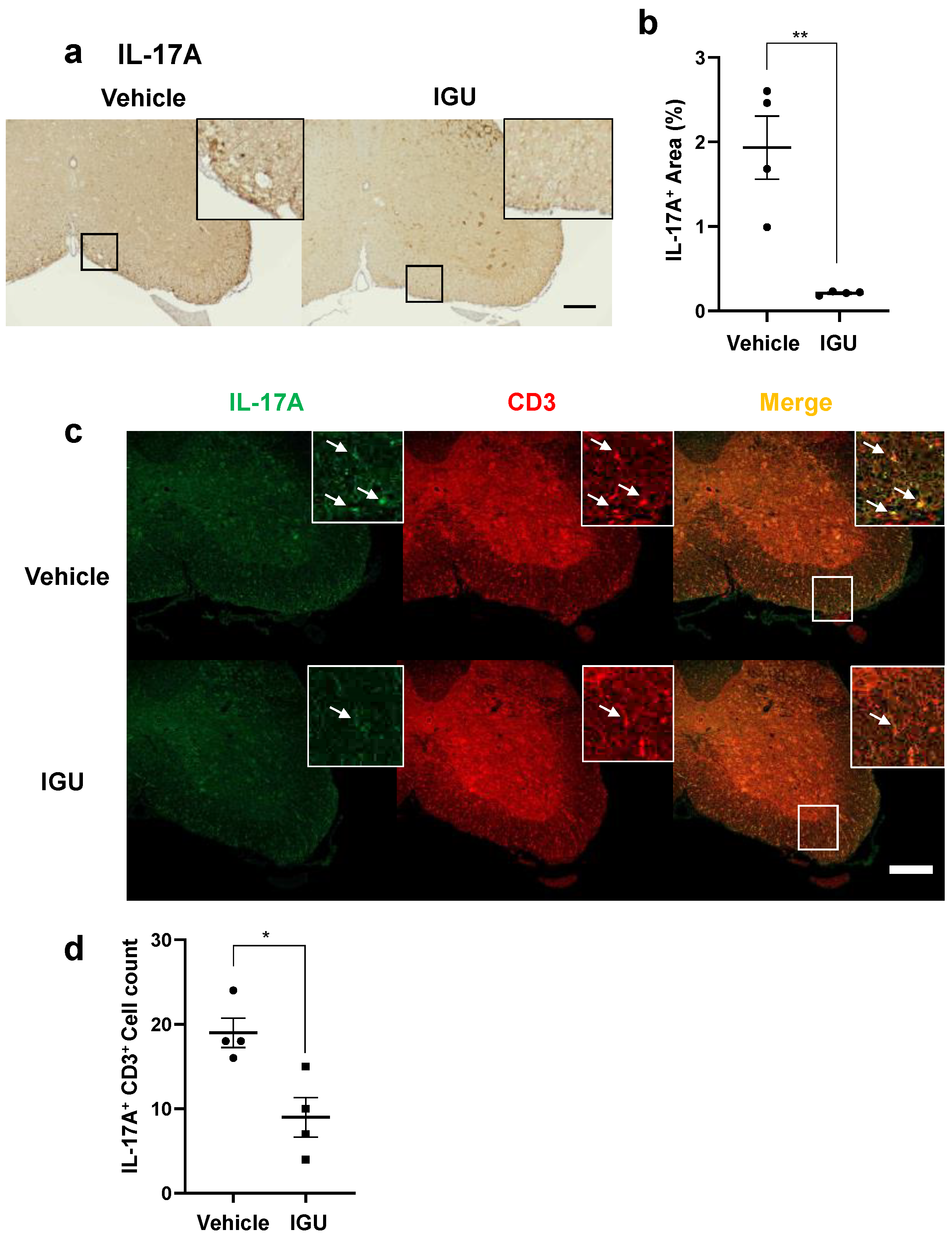
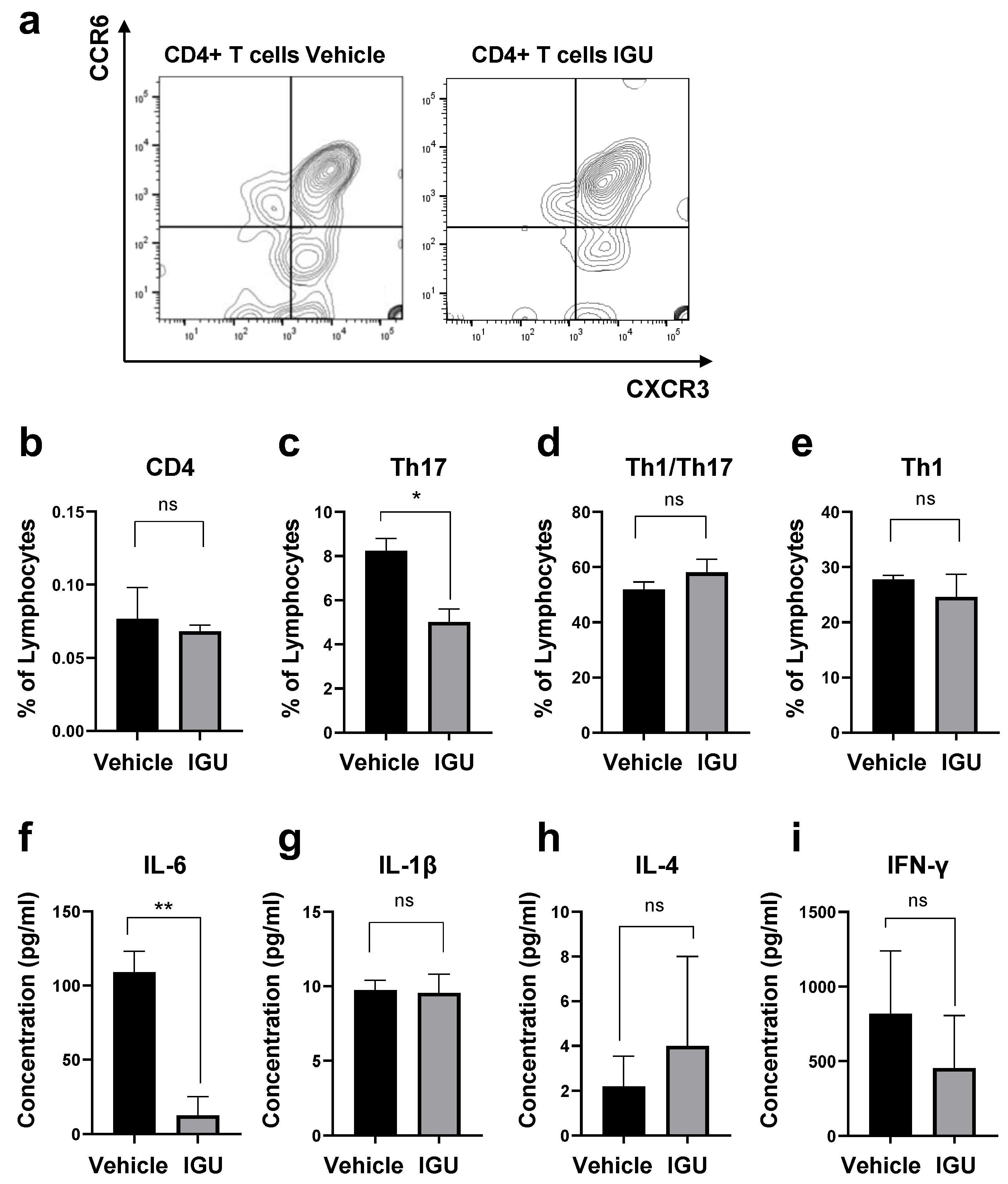
3.4. IGU Suppressed Th17 Cell Migration in the Spinal Cord and Cerebrospinal Fluid (CSF) IL-6 Production during the Chronic Phase
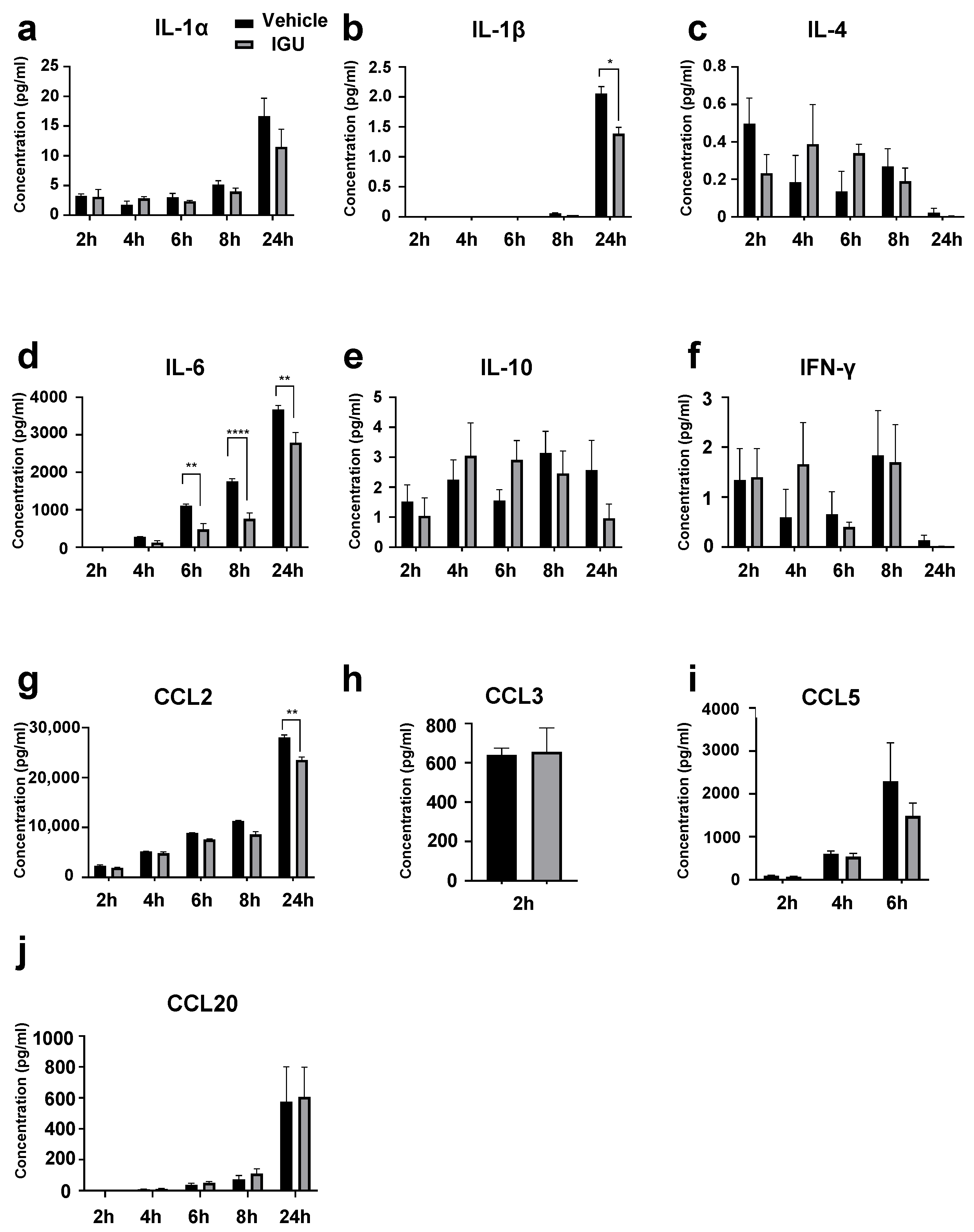
3.5. IGU Inhibited IL-6 and CCL2 Release from Glial Cells in the In Vitro Activated Glial Inflammation Model
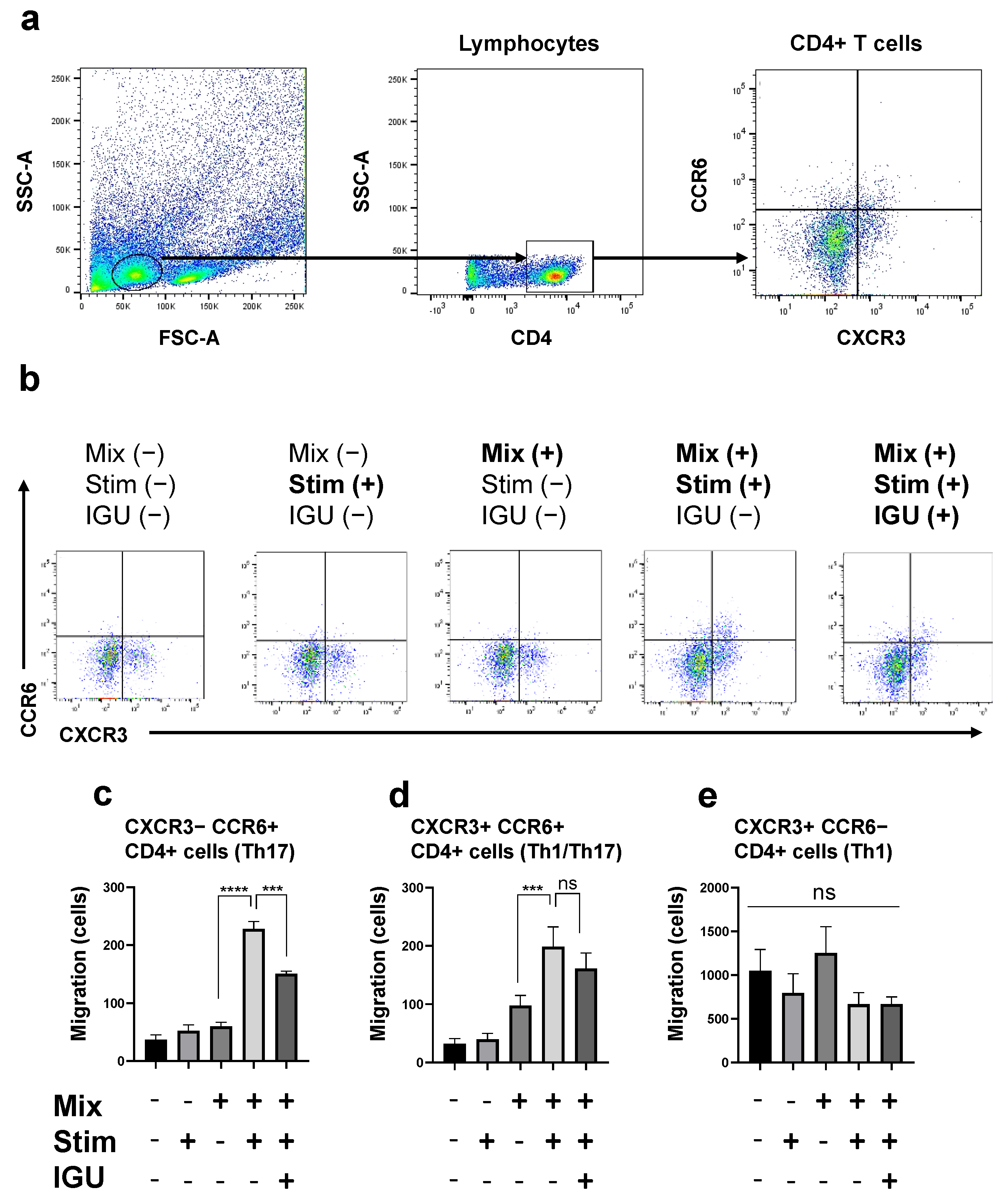
3.6. IGU Decreased the Number of Th17 Cells Migrating toward the Glial Cell Culture in the In Vitro Migration Assay
3.7. IGU Did Not Affect the Differentiation of CD4+ T Cells or T Cell Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tillery, E.E.; Clements, J.N.; Howard, Z. What’s New in Multiple Sclerosis? Ment. Health Clin. 2018, 7, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Dermietzel, R.; Farooq, M.; Kessler, J.A.; Althaus, H.; Hertzberg, E.L.; Spray, D.C. Oligodendrocytes express gap junction proteins connexin32 and connexin45. Glia 1997, 20, 101–114. [Google Scholar] [CrossRef]
- Kunzelmann, P.; Blumcke, I.; Traub, O.; Dermietzel, R.; Willecke, K. Coexpression of connexin45 and -32 in oligodendrocyte of rat brain. J. Neurocytol. 1997, 26, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Altevogt, B.M.; Kleopa, K.A.; Postma, F.R.; Scherer, S.S.; Paul, D.L. Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 2002, 22, 6458–6470. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Connexin29 and connexin32 at oligodendrocyte/astrocyte gap junctions and in myelin of mouse CNS. J. Comp. Neurol. 2003, 464, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Dudek, F.; Rash, J.E. Update on connexins and gap junctions in neurons and glia in the nervous system. Brain Res. Rev. 2004, 47, 191–215. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ochalski, A.; Hertzberg, E.; Nagy, J. LM and EM immunolocalization of the gap junctions protein connexin43 in rat brain. Brain Res. 1990, 508, 313–319. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ochalski, A.; Hertzberg, E.L.; Nagy, J.I. On the organization of astrocytic gap junctions in rat brain as suggested by LM and EM immunohistochemistry of connexin43 expression. J. Comp. Neurol. 1990, 302, 853–883. [Google Scholar] [CrossRef]
- Nagy, J.; Ochalski, P.; Li, J.; Hertzberg, E. Evidence for co-localization of another connexin-43 at astrocytic gap junctions in rat brain. Neuroscience 1997, 78, 533–548. [Google Scholar] [CrossRef]
- Nagy, J.I.; Patel, D.; Ochalski, P.A.Y.; Stelmack, G. Connexin30 in rodent, cat and human brain: Selective expression in gray matter astrocytes, co-localization with connexin30 at gap junctions and late developmental appearance. Neuroscience 1999, 88, 447–468. [Google Scholar] [CrossRef]
- Nagy, J.I.; Li, X.; Rempel, J.; Stelmack, G.; Patel, D.; Staines, W.A.; Yasumura, T.; Rash, J.E. Connexin26 in adult rodent central nervous system: Demonstration at astrocytic gap junctions and colocalization with connexin30 and connexin43. J. Comp. Neurol. 2001, 441, 302–323. [Google Scholar] [CrossRef] [PubMed]
- Masaki, K.; Suzuki, S.O.; Matsushita, T.; Matsuoka, T.; Imamura, S.; Yamasaki, R.; Suzuki, M.; Suenaga, T.; Iwaki, T.; Kira, J.-I. Connexin 43 astrocytopathy linked to rapidly progressive multiple sclerosis and neuromyelitis optica. PLoS ONE 2013, 8, e72919. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yamasaki, R.; Yamaguchi, H.; Nagata, S.; Une, H.; Cui, Y.; Masaki, K.; Nakamuta, Y.; Iinuma, K.; Watanabe, M.; et al. Oligodendroglial connexin 47 regulates neuroinflammation upon autoimmune demyelination in a novel mouse model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2020, 117, 2160–2169. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kawasaki, H.; Kurata, K.; Aikawa, Y.; Tsukamoto, Y.; Inaba, T. T-614, a novel antirheumatic drug, inhibits both the activity and induction of cyclooxygenase-2 (COX-2) in cultured fibroblasts. J. Pharmacol. 1995, 67, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Aikawa, Y.; Tsubouchi, Y.; Hashiramoto, A.; Yamada, R.; Kawahito, Y.; Inoue, K.; Kusaka, Y.; Kondo, M.; Sano, H. Inhibitory effect of T-614 on tumor necrosis factor-α induced cytokine production and nuclear factor-κB activation in cultured human synovial cells. J. Reumatol. 2001, 28, 2591–2596. [Google Scholar]
- Aikawa, Y.; Yamamoto, M.; Yamamoto, T.; Morimoto, K.; Tanaka, K. An anti-rheumatic agent T-614 inhibits NF-κB activation in LPS-and TNF-α-stimulated THP-1 cells without interfering with IκBα degradation. Inflamm. Res. 2002, 51, 188–194. [Google Scholar] [CrossRef]
- Bloom, J.; Metz, C.; Nalawade, S.; Casabar, J.; Cheng, K.F.; He, M.; Sherry, B.; Coleman, T.; Forsthuber, T.; Al-Abed, Y. Identification of iguratimod as an inhibitor of macrophage migration inhibitory factor (MIF) with steroid-sparing potential. J. Biol. Chem. 2016, 291, 26502–26514. [Google Scholar] [CrossRef]
- Ye, Y.; Liu, M.; Tang, L.; Du, F.; Liu, Y.; Hao, P.; Fu, Q.; Guo, Q.; Yan, Q.; Zhang, X.; et al. Iguratimod represses B cell terminal differentiation linked with the inhibition of PKC/EGR1 axis. Arthritis Res. Ther. 2019, 21, 92. [Google Scholar] [CrossRef]
- Li, G.; Yamasaki, R.; Fang, M.; Masaki, K.; Ochi, H.; Matsushita, T.; Kira, J.I. Novel disease-modifying anti-rheumatic drug iguratimod suppresses chronic experimental autoimmune encephalomyelitis by down-regulation activation of macrophages/microglia through an NF-κB pathway. Sci. Rep. 2018, 8, 1933. [Google Scholar] [CrossRef] [PubMed]
- Karaki, H. Guidelines for Proper Conduct of Animal Experiments by the Science Council of Japan. Altern. Anim. Test. Exp. 2020, 25, 72. [Google Scholar]
- Du Sert, N.P.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; Emerson, M.; et al. Reporting animal research: Explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 2020, 18, 183–188. [Google Scholar]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef]
- Mishina, M.; Sakimura, K. Conditional gene targeting on the pure C57BL/6 genetic background. Neurosci. Res. 2007, 58, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, N.H.; Macklin, W.B.; Popko, B. Inducible site-specific recombination in myelination cells. Genesis 2003, 35, 63–72. [Google Scholar] [CrossRef]
- Schwenk, F.; Kühn, R.; Rajewsky, K.; Angrand, P.-O.; Stewart, A.F. Temporally and spatially regulated somatic mutagenesis in mice. Nucleic Acids Res. 1998, 26, 1427–1432. [Google Scholar] [CrossRef]
- Yamasaki, R.; Fujii, T.; Wang, B.; Masaki, K.; Kido, M.A.; Yoshida, M.; Matsushita, T.; Kira, J.-I. Allergic inflammation leads to neuropathic pain via glial cell activation. J. Neurosci. 2016, 36, 11929–11945. [Google Scholar] [CrossRef]
- Wight, P.A.; Duchala, C.S.; Readhead, C.; Macklin, W.B. A myelin proteolipid protein-LacZ fusion protein is developmentally regulated and targeted to the myelin membrane in transgenic mice. J. Cell Biol. 1993, 123, 443–454. [Google Scholar] [CrossRef]
- Gosselin, A.; Monteiro, P.; Chomont, N.; Diaz-Griffero, F.; Said, E.A.; Fonseca, S.; Wacleche, V.; El-Far, M.; Boulassel, M.-R.; Routy, J.-P.; et al. Peripheral blood CCR4+CCR6+ and CXCR3+CCR6+ CD4+ T cells are highly permissive to HIV-1 infection. J. Immunol. 2010, 184, 1604–1616. [Google Scholar] [CrossRef]
- Suzumura, A.; Bhat, S.; Eccleston, P.A.; Lisak, R.P.; Silberberg, D.H. The isolation and long-term culture of oligodendrocytes from newborn mouse brain. Brain Res. 1984, 324, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Masaki, K.; Yamasaki, R.; Kawanokuchi, J.; Takeuchi, H.; Matsushita, T.; Suzumura, A.; Kira, J.-I. Th1 cells downregulate connexin 43 gap junctions in astrocytes via microglial activation. Sci. Rep. 2016, 6, 38387. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, A.E.; Salm, A.K. Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. J. Neurosci. 2003, 23, 7759–7766. [Google Scholar] [CrossRef]
- Mimori, T.; Harigai, M.; Atsumi, T.; Fujii, T.; Kuwana, M.; Matsuno, H.; Momohara, S.; Takei, S.; Tamura, N.; Takasaki, Y.; et al. Safety and effectiveness of iguratimod in patients with rheumatoid arthritis: Final report of a 52-week, multicenter postmarketing surveillance study. Mod. Rheumatol. 2019, 29, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, Y.; Tanuma, N.; Shin, T.; Makino, S.; Tanaka, K.; Matsumoto, Y. A new anti-rheumatic drug, T-614, effectively suppresses the development of autoimmune encephalomyelitis. J. Neuroimmunol. 1998, 89, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef]
- Morimoto, K.; Miura, A.; Tanaka, K. Anti-allodynic action of the disease-modifying anti-rheumatic drug iguratimod in a rat model of neuropathic pain. Inflamm. Res. 2017, 66, 855–862. [Google Scholar] [CrossRef]
- Korn, T.; Reddy, J.; Gao, W.; Bettelli, E.; Awasthi, A.; Petersen, T.R.; Bäckström, B.T.; Sobel, R.A.; Wucherpfennig, K.W.; Strom, T.B.; et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat. Med. 2007, 13, 423–431. [Google Scholar] [CrossRef]
- Rostami, A.; Ciric, B. Role of Th17 cells in the pathogenesis of CNS inflammatory demyelination. J. Neurol. Sci. 2013, 333, 76–87. [Google Scholar] [CrossRef]
- Brucklacher-Waldert, V.; Stuerner, K.; Kolster, M.; Wolthausen, J.; Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 2009, 132, 3329–3341. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.K.; Wang, L.; Han, P.; Zhang, X.; Ekholm, S.; Srinivasan, A.; Irani, D.N.; Segal, B.M. Dysregulation of the IL-23/IL-17 axis and myeloid factors in secondary progressive MS. Neurology 2014, 83, 1500–1507. [Google Scholar] [CrossRef]
- Hirota, K.; Yoshitomi, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Aranami, T.; Yamamura, T. Human Th17 cells are identified as bearing CCR2+ CCR5- phenotype. J. Immunol. 2007, 178, 7525–7529. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, N.J.; Benveniste, E.N. Interleukin-6 expression and regulation in astrocytes. J. Neuroimmunol. 1999, 100, 124–139. [Google Scholar] [CrossRef]
- Ringheim, G.E.; Burgher, K.L.; Heroux, J.A. Interleukin-6 mRNA expression by cortical neurons in culture: Evidence for neuronal sources of interleukin-6 production in the brain. J. Neuroimmunol. 1995, 63, 113–123. [Google Scholar] [CrossRef]
- Krasowska-Zoladek, A.; Banaszewska, M.; Kraszpulski, M.; Konat, G.W. Kinetics of inflammatory response of astrocytes induced by TLR3 and TLR4 ligation. J. Neurosci. 2007, 85, 205–212. [Google Scholar]
- Lu, Y.; Li, B.; Xu, A.; Liang, X.; Xu, T.; Jin, H.; Xie, Y.; Wang, R.; Liu, X.; Gao, X.; et al. NF-κB and AP-1 are required for the lipopolysaccharide-induced expression of MCP-1, CXCL1, and Cx43 in cultured rat dorsal spinal cord astrocytes. Front. Mol. Neurosci. 2022, 15, 859558. [Google Scholar] [CrossRef]
- Takami, S.; Nishikawa, H.; Minami, M.; Nishiyori, A.; Sato, M.; Akaike, A.; Satoh, M. Induction of macrophage inflammatory protein MIP-1α mRNA on glial cells after focal cerebral ischemia in the rat. Neurosci. Lett. 1997, 227, 173–176. [Google Scholar] [CrossRef]
- Orthmann-Murphy, J.L.; Abrams, C.K.; Scherer, S.S. Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 2008, 35, 101–116. [Google Scholar] [CrossRef]
- Takeuchi, H.; Suzumura, A. Gap junctions and hemichannels composed of connexins: Potential therapeutic targets for neurodegenerative diseases. Front. Cell. Neurosci. 2014, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Murphy, Á.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagata, S.; Yamasaki, R.; Takase, E.O.; Iida, K.; Watanabe, M.; Masaki, K.; Wijering, M.H.C.; Yamaguchi, H.; Kira, J.-i.; Isobe, N. Iguratimod Ameliorates the Severity of Secondary Progressive Multiple Sclerosis in Model Mice by Directly Inhibiting IL-6 Production and Th17 Cell Migration via Mitigation of Glial Inflammation. Biology 2023, 12, 1217. https://doi.org/10.3390/biology12091217
Nagata S, Yamasaki R, Takase EO, Iida K, Watanabe M, Masaki K, Wijering MHC, Yamaguchi H, Kira J-i, Isobe N. Iguratimod Ameliorates the Severity of Secondary Progressive Multiple Sclerosis in Model Mice by Directly Inhibiting IL-6 Production and Th17 Cell Migration via Mitigation of Glial Inflammation. Biology. 2023; 12(9):1217. https://doi.org/10.3390/biology12091217
Chicago/Turabian StyleNagata, Satoshi, Ryo Yamasaki, Ezgi Ozdemir Takase, Kotaro Iida, Mitsuru Watanabe, Katsuhisa Masaki, Marion Heleen Cathérine Wijering, Hiroo Yamaguchi, Jun-ichi Kira, and Noriko Isobe. 2023. "Iguratimod Ameliorates the Severity of Secondary Progressive Multiple Sclerosis in Model Mice by Directly Inhibiting IL-6 Production and Th17 Cell Migration via Mitigation of Glial Inflammation" Biology 12, no. 9: 1217. https://doi.org/10.3390/biology12091217