

Using i-GONAD for Cell-Type-Specific and Systematic Analysis of Developmental Transcription Factors In Vivo
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. CRISPR/Cas9
2.2. i-GONAD
2.3. Genotyping and Sanger Sequencing
2.4. Immunohistochemistry
2.5. Fluorescent-Activated Cell Sorting
2.6. Western Blot
3. Results
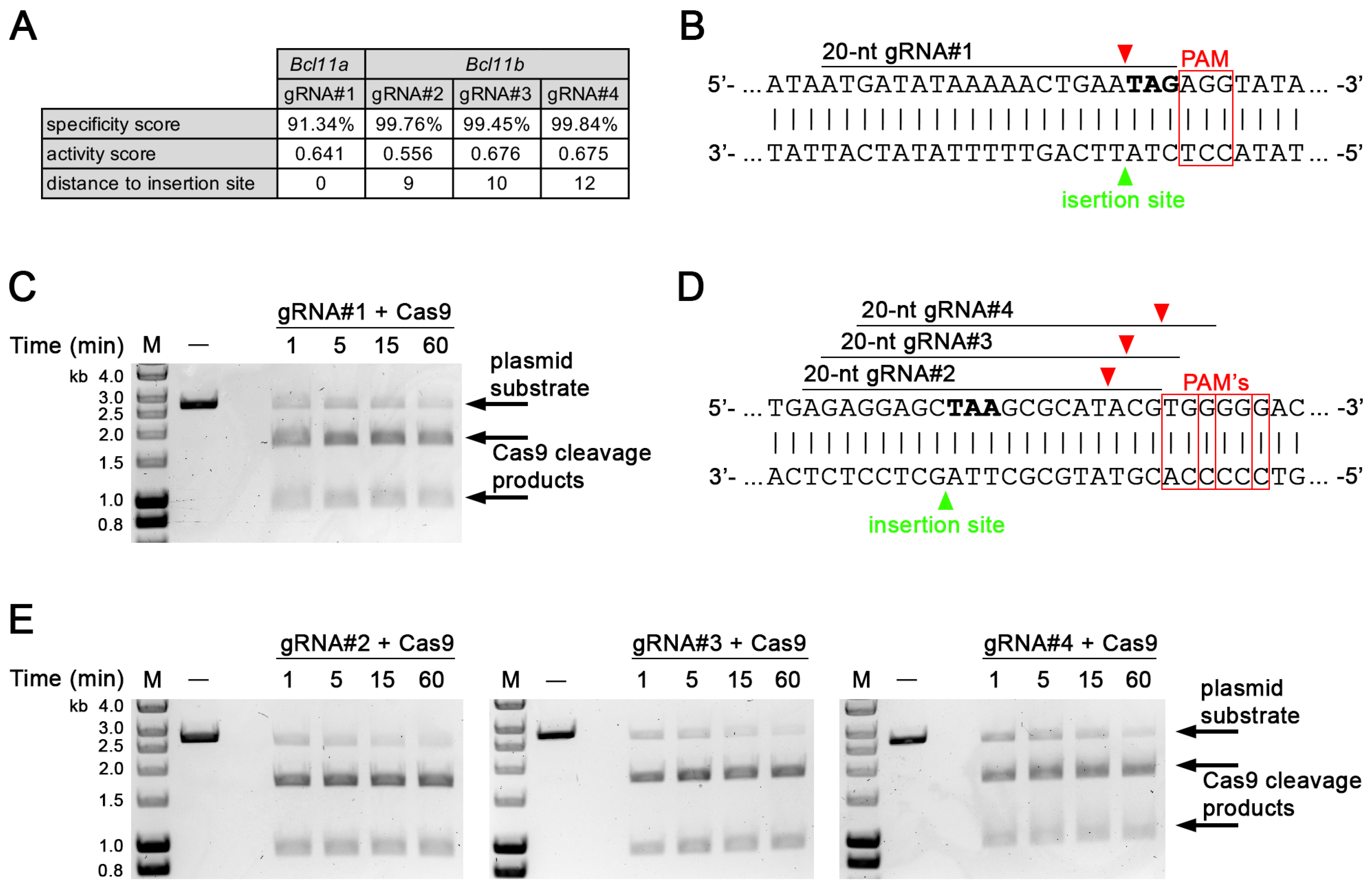
3.1. Identification and In Vitro Validation of gRNA Targeting in Bcl11a and Bcl11b
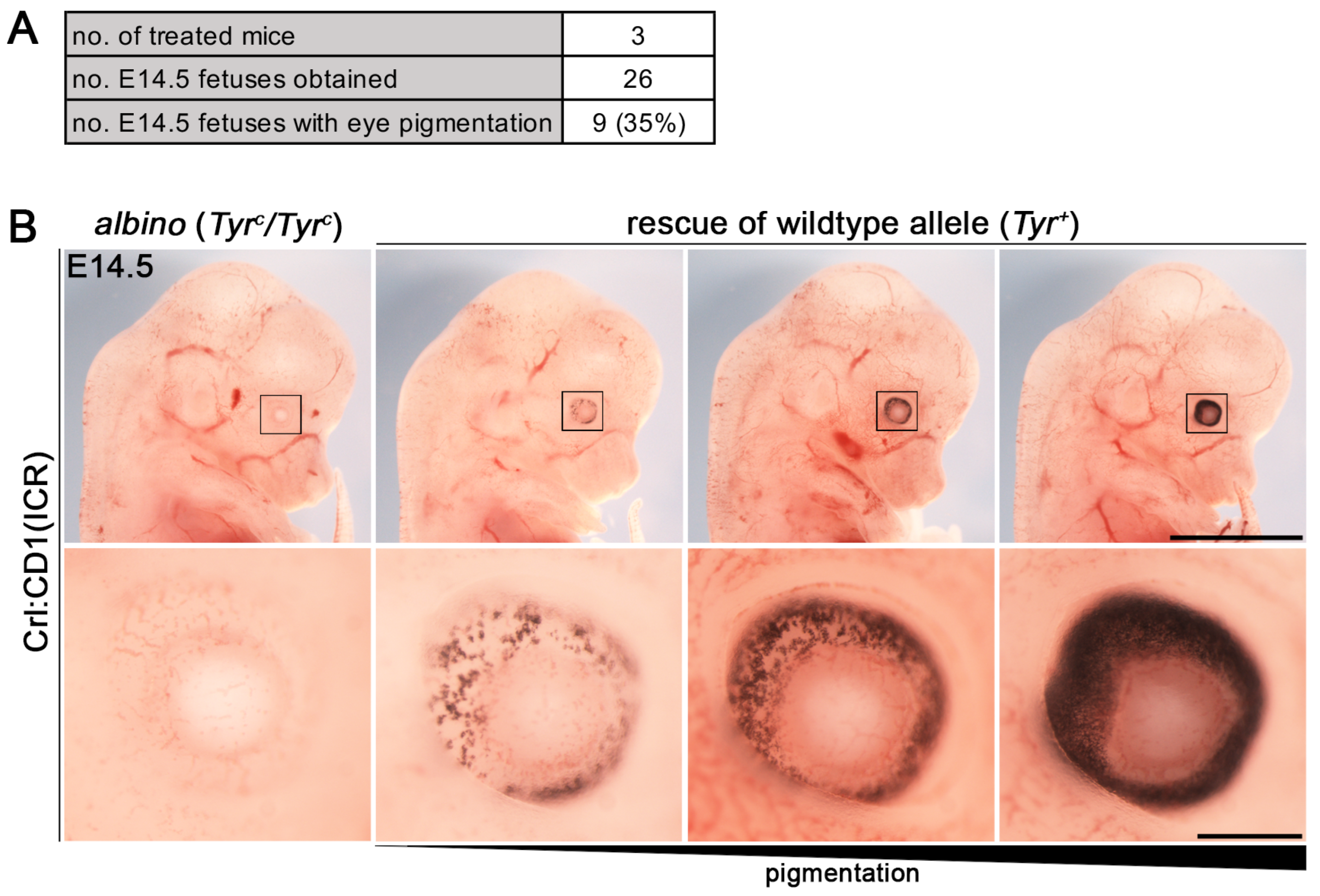
3.2. Establishment of i-GONAD Method
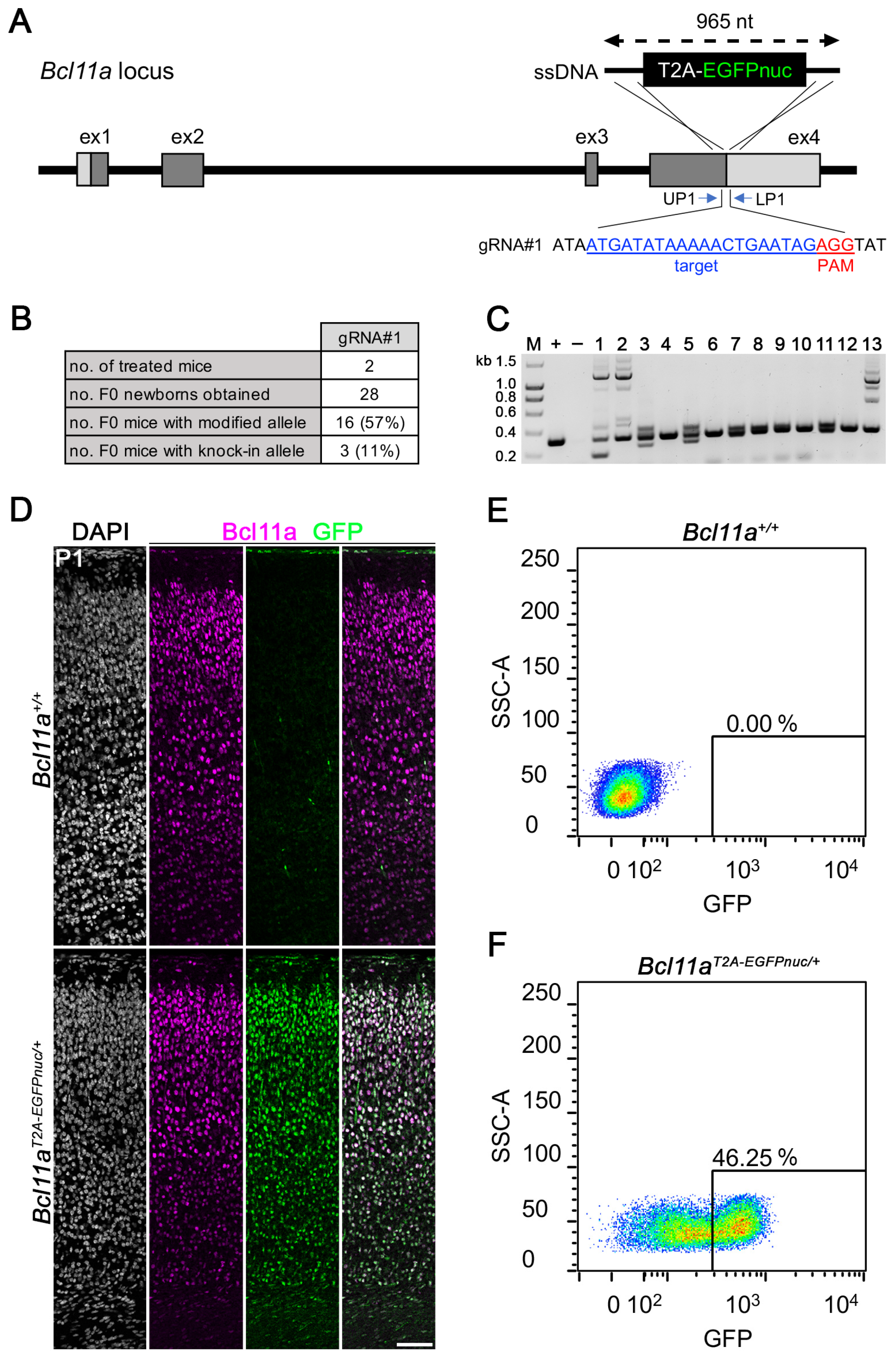
3.3. Generation of Bcl11aT2A-EGFPnuc Allele Using i-GONAD
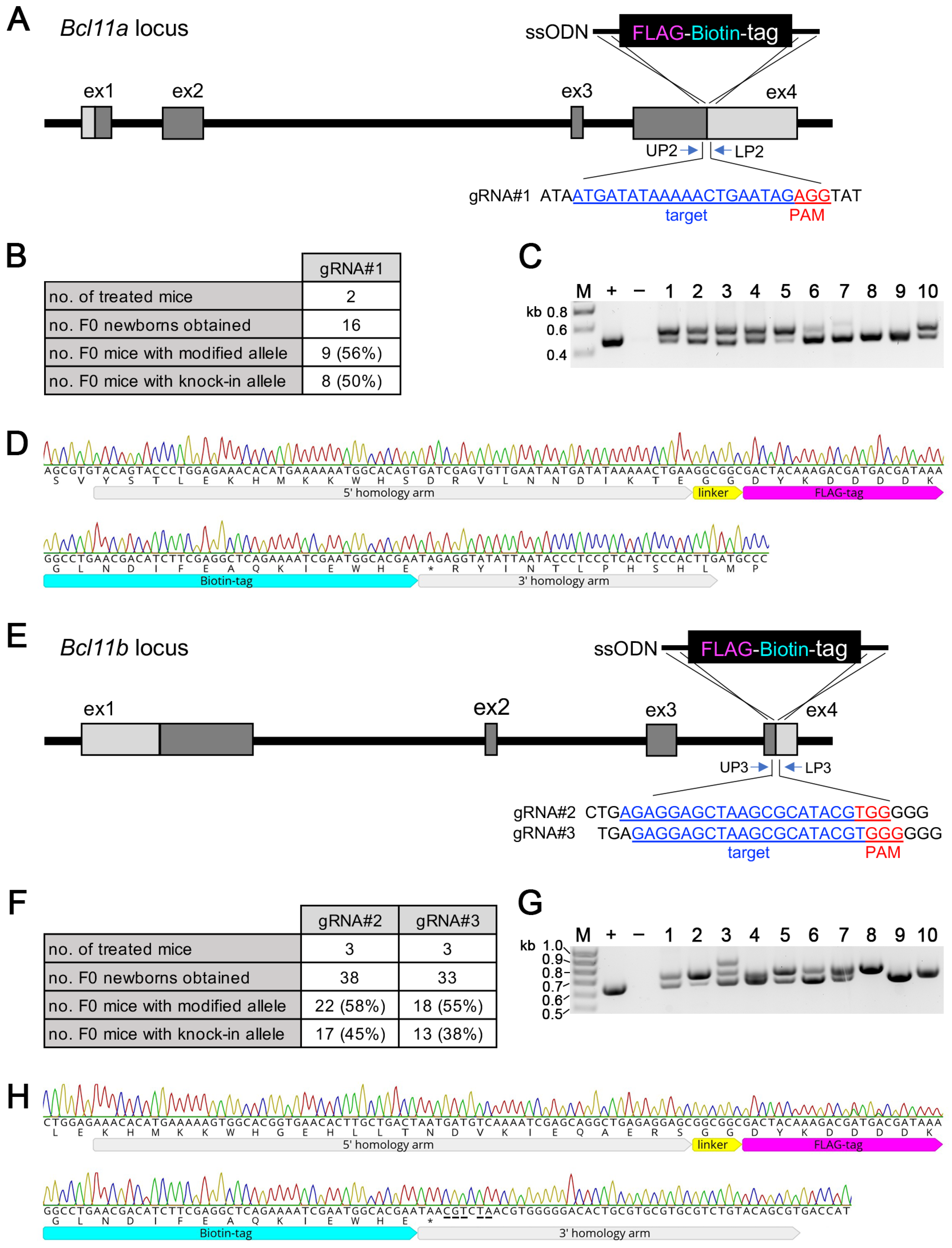
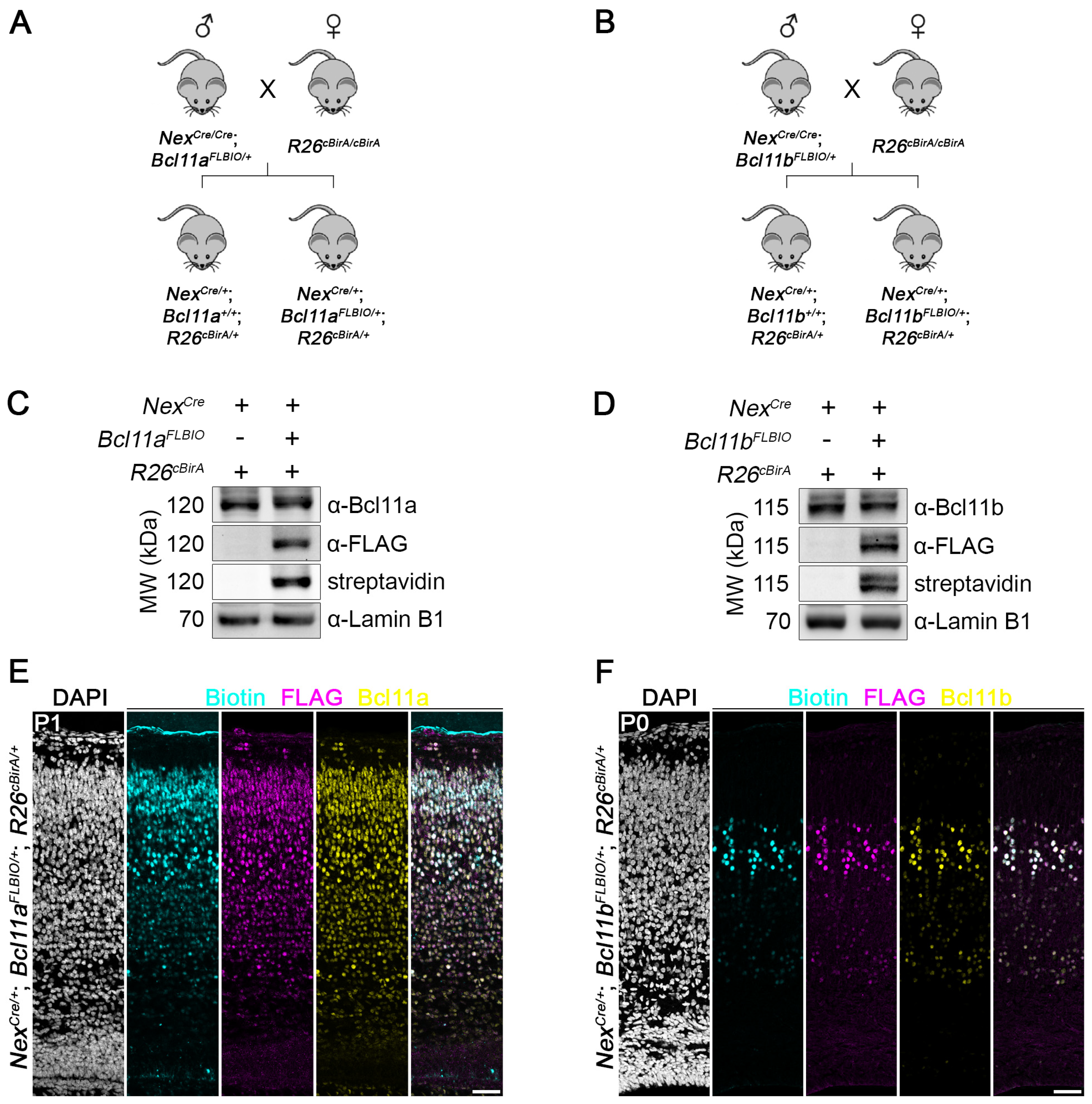
3.4. Generation of Bcl11aFLBIO and Bcl11bFLBIO Alleles Using i-GONAD
3.5. Efficient In Vivo Biotinylation of FLBIO-Tagged Bcl11a and Bcl11b
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Ravasi, T.; Suzuki, H.; Cannistraci, C.V.; Katayama, S.; Bajic, V.B.; Tan, K.; Akalin, A.; Schmeier, S.; Kanamori-Katayama, M.; Bertin, N.; et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 2010, 140, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, M.; Sato, M.; Miura, H.; Takabayashi, S.; Matsuyama, M.; Koyano, T.; Arifin, N.; Nakamura, S.; Wada, K.; Gurumurthy, C.B. i-GONAD: A robust method for in situ germline genome engineering using CRISPR nucleases. Genome Biol. 2018, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Quadros, R.M.; Miura, H.; Harms, D.W.; Akatsuka, H.; Sato, T.; Aida, T.; Redder, R.; Richardson, G.P.; Inagaki, Y.; Sakai, D.; et al. Easi-CRISPR: A robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 2017, 18, 92. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Takabayashi, S.; Mutoh, H.; Saitsu, H. Generation of Flag/DYKDDDDK Epitope Tag Knock-In Mice Using i-GONAD Enables Detection of Endogenous CaMKIIalpha and beta Proteins. Int. J. Mol. Sci. 2022, 23, 11915. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.M.; Mutoh, H.; Aoto, K.; Belal, H.; Saitsu, H. Cnpy3(2xHA) mice reveal neuronal expression of Cnpy3 in the brain. J. Neurosci. Methods 2023, 383, 109730. [Google Scholar] [CrossRef] [PubMed]
- Nakano, H.; Kawai, S.; Ooki, Y.; Chiba, T.; Ishii, C.; Nozawa, T.; Utsuki, H.; Umemura, M.; Takahashi, S.; Takahashi, Y. Functional validation of epitope-tagged ATF5 knock-in mice generated by improved genome editing of oviductal nucleic acid delivery (i-GONAD). Cell Tissue Res. 2021, 385, 239–249. [Google Scholar] [CrossRef]
- de Boer, E.; Rodriguez, P.; Bonte, E.; Krijgsveld, J.; Katsantoni, E.; Heck, A.; Grosveld, F.; Strouboulis, J. Efficient biotinylation and single-step purification of tagged transcription factors in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 2003, 100, 7480–7485. [Google Scholar] [CrossRef]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef]
- Kim, J.; Chu, J.; Shen, X.; Wang, J.; Orkin, S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell 2008, 132, 1049–1061. [Google Scholar] [CrossRef]
- Simon, R.; Wiegreffe, C.; Britsch, S. Bcl11 Transcription Factors Regulate Cortical Development and Function. Front. Mol. Neurosci. 2020, 13, 51. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.; Estruch, S.B.; Graham, S.A.; McRae, J.; Sawiak, S.J.; Hurst, J.A.; Joss, S.K.; Holder, S.E.; Morton, J.E.; Turner, C.; et al. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am. J. Hum. Genet. 2016, 99, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Lessel, D.; Gehbauer, C.; Bramswig, N.C.; Schluth-Bolard, C.; Venkataramanappa, S.; van Gassen, K.L.I.; Hempel, M.; Haack, T.B.; Baresic, A.; Genetti, C.A.; et al. BCL11B mutations in patients affected by a neurodevelopmental disorder with reduced type 2 innate lymphoid cells. Brain 2018, 141, 2299–2311. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.; Brylka, H.; Schwegler, H.; Venkataramanappa, S.; Andratschke, J.; Wiegreffe, C.; Liu, P.; Fuchs, E.; Jenkins, N.A.; Copeland, N.G.; et al. A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J. 2012, 31, 2922–2936. [Google Scholar] [CrossRef]
- Wiegreffe, C.; Simon, R.; Peschkes, K.; Kling, C.; Strehle, M.; Cheng, J.; Srivatsa, S.; Liu, P.; Jenkins, N.A.; Copeland, N.G.; et al. Bcl11a (Ctip1) Controls Migration of Cortical Projection Neurons through Regulation of Sema3c. Neuron 2015, 87, 311–325. [Google Scholar] [CrossRef]
- Wiegreffe, C.; Wahl, T.; Joos, N.S.; Bonnefont, J.; Liu, P.; Britsch, S. Developmental cell death of cortical projection neurons is controlled by a Bcl11a/Bcl6-dependent pathway. EMBO Rep. 2022, 23, e54104. [Google Scholar] [CrossRef]
- Cismasiu, V.B.; Adamo, K.; Gecewicz, J.; Duque, J.; Lin, Q.; Avram, D. BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene 2005, 24, 6753–6764. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Anders, C.; Jinek, M. In vitro enzymology of Cas9. Methods Enzymol. 2014, 546, 1–20. [Google Scholar] [CrossRef]
- Bollen, Y.; Post, J.; Koo, B.K.; Snippert, H.J.G. How to create state-of-the-art genetic model systems: Strategies for optimal CRISPR-mediated genome editing. Nucleic Acids Res. 2018, 46, 6435–6454. [Google Scholar] [CrossRef]
- Miura, H.; Quadros, R.M.; Gurumurthy, C.B.; Ohtsuka, M. Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat. Protoc. 2018, 13, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Gurumurthy, C.B.; Sato, M.; Nakamura, A.; Inui, M.; Kawano, N.; Islam, M.A.; Ogiwara, S.; Takabayashi, S.; Matsuyama, M.; Nakagawa, S.; et al. Creation of CRISPR-based germline-genome-engineered mice without ex vivo handling of zygotes by i-GONAD. Nat. Protoc. 2019, 14, 2452–2482. [Google Scholar] [CrossRef] [PubMed]
- Byers, S.L.; Wiles, M.V.; Dunn, S.L.; Taft, R.A. Mouse estrous cycle identification tool and images. PLoS ONE 2012, 7, e35538. [Google Scholar] [CrossRef] [PubMed]
- Goebbels, S.; Bormuth, I.; Bode, U.; Hermanson, O.; Schwab, M.H.; Nave, K.A. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 2006, 44, 611–621. [Google Scholar] [CrossRef]
- Wood, K.H.; Johnson, B.S.; Welsh, S.A.; Lee, J.Y.; Cui, Y.; Krizman, E.; Brodkin, E.S.; Blendy, J.A.; Robinson, M.B.; Bartolomei, M.S.; et al. Tagging methyl-CpG-binding domain proteins reveals different spatiotemporal expression and supports distinct functions. Epigenomics 2016, 8, 455–473. [Google Scholar] [CrossRef]
- John, A.; Brylka, H.; Wiegreffe, C.; Simon, R.; Liu, P.; Juttner, R.; Crenshaw, E.B., 3rd; Luyten, F.P.; Jenkins, N.A.; Copeland, N.G.; et al. Bcl11a is required for neuronal morphogenesis and sensory circuit formation in dorsal spinal cord development. Development 2012, 139, 1831–1841. [Google Scholar] [CrossRef]
- Grandi, F.C.; Modi, H.; Kampman, L.; Corces, M.R. Chromatin accessibility profiling by ATAC-seq. Nat. Protoc. 2022, 17, 1518–1552. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Kim, J.; Cantor, A.B.; Orkin, S.H.; Wang, J. Use of in vivo biotinylation to study protein-protein and protein-DNA interactions in mouse embryonic stem cells. Nat. Protoc. 2009, 4, 506–517. [Google Scholar] [CrossRef]
- Tolve, M.; Ulusoy, A.; Patikas, N.; Islam, K.U.S.; Bodea, G.O.; Ozturk, E.; Broske, B.; Mentani, A.; Wagener, A.; van Loo, K.M.J.; et al. The transcription factor BCL11A defines distinct subsets of midbrain dopaminergic neurons. Cell Rep. 2021, 36, 109697. [Google Scholar] [CrossRef] [PubMed]
- Marshall, O.J.; Southall, T.D.; Cheetham, S.W.; Brand, A.H. Cell-type-specific profiling of protein-DNA interactions without cell isolation using targeted DamID with next-generation sequencing. Nat. Protoc. 2016, 11, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Mann, M. Protein interaction screening by quantitative immunoprecipitation combined with knockdown (QUICK). Nat. Methods 2006, 3, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.J.; Peric-Hupkes, D.; van Steensel, B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat. Protoc. 2007, 2, 1467–1478. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiegreffe, C.; Ehricke, S.; Schmid, L.; Andratschke, J.; Britsch, S. Using i-GONAD for Cell-Type-Specific and Systematic Analysis of Developmental Transcription Factors In Vivo. Biology 2023, 12, 1236. https://doi.org/10.3390/biology12091236
Wiegreffe C, Ehricke S, Schmid L, Andratschke J, Britsch S. Using i-GONAD for Cell-Type-Specific and Systematic Analysis of Developmental Transcription Factors In Vivo. Biology. 2023; 12(9):1236. https://doi.org/10.3390/biology12091236
Chicago/Turabian StyleWiegreffe, Christoph, Simon Ehricke, Luisa Schmid, Jacqueline Andratschke, and Stefan Britsch. 2023. "Using i-GONAD for Cell-Type-Specific and Systematic Analysis of Developmental Transcription Factors In Vivo" Biology 12, no. 9: 1236. https://doi.org/10.3390/biology12091236