
Amphiregulin Upregulation in Visfatin-Stimulated Colorectal Cancer Cells Reduces Sensitivity to 5-Fluororacil Cytotoxicity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Real-Time PCR
2.4. Western Blot
2.5. MTT Assay
2.6. Statistical Analysis
3. Results
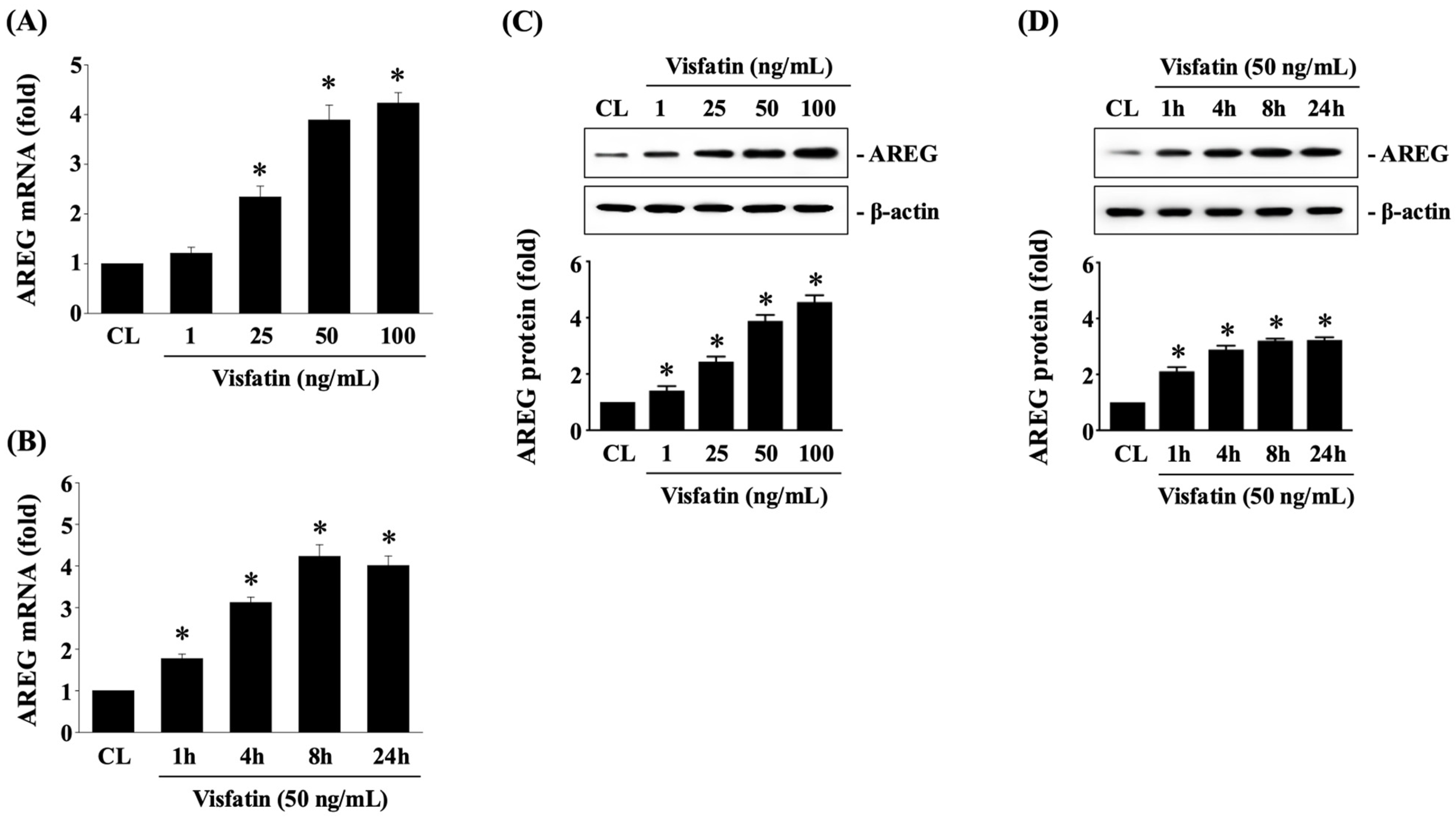
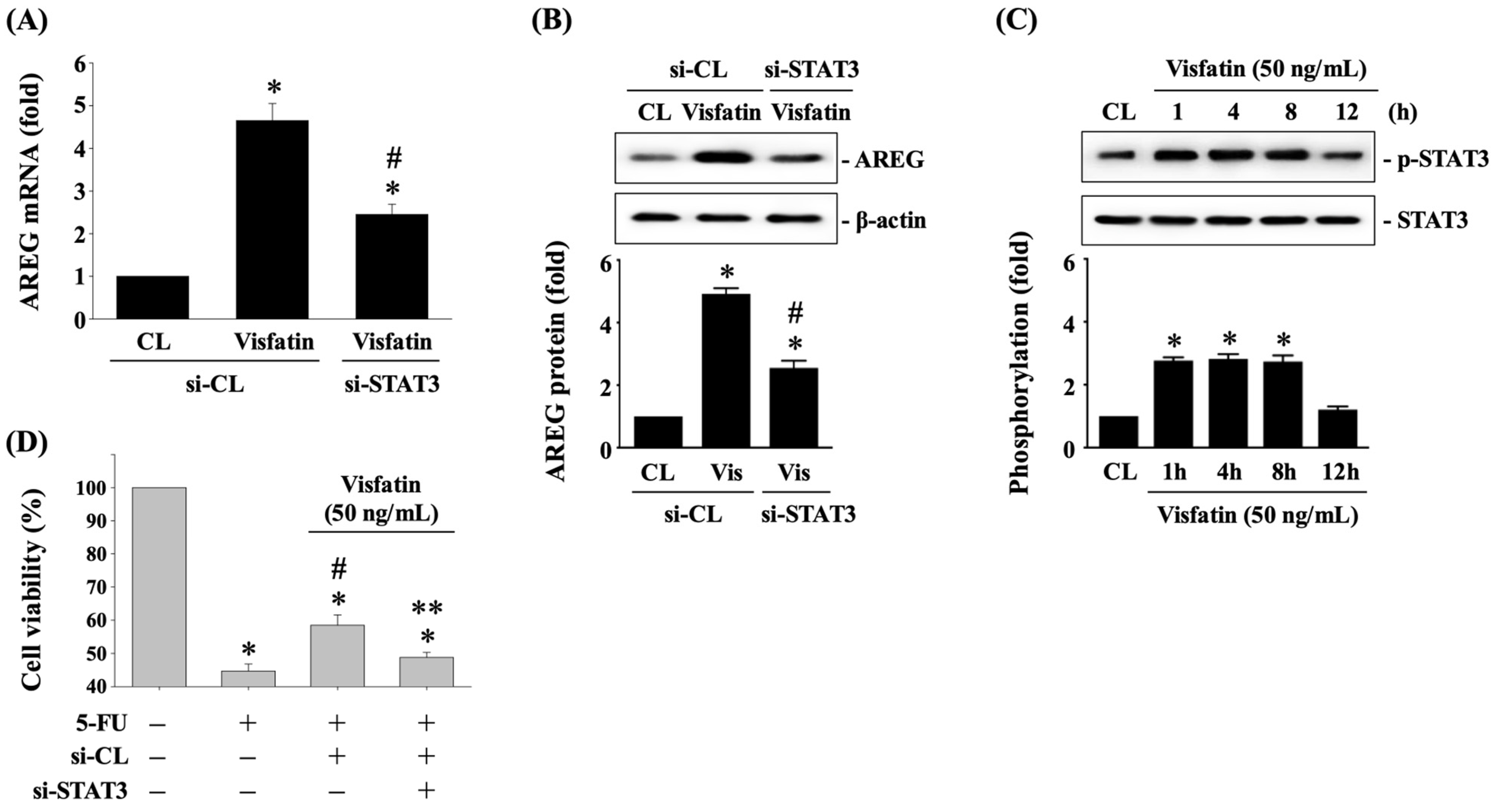
3.1. Visfatin Upregulates AREG Expression in HCT-116 CRC Cells
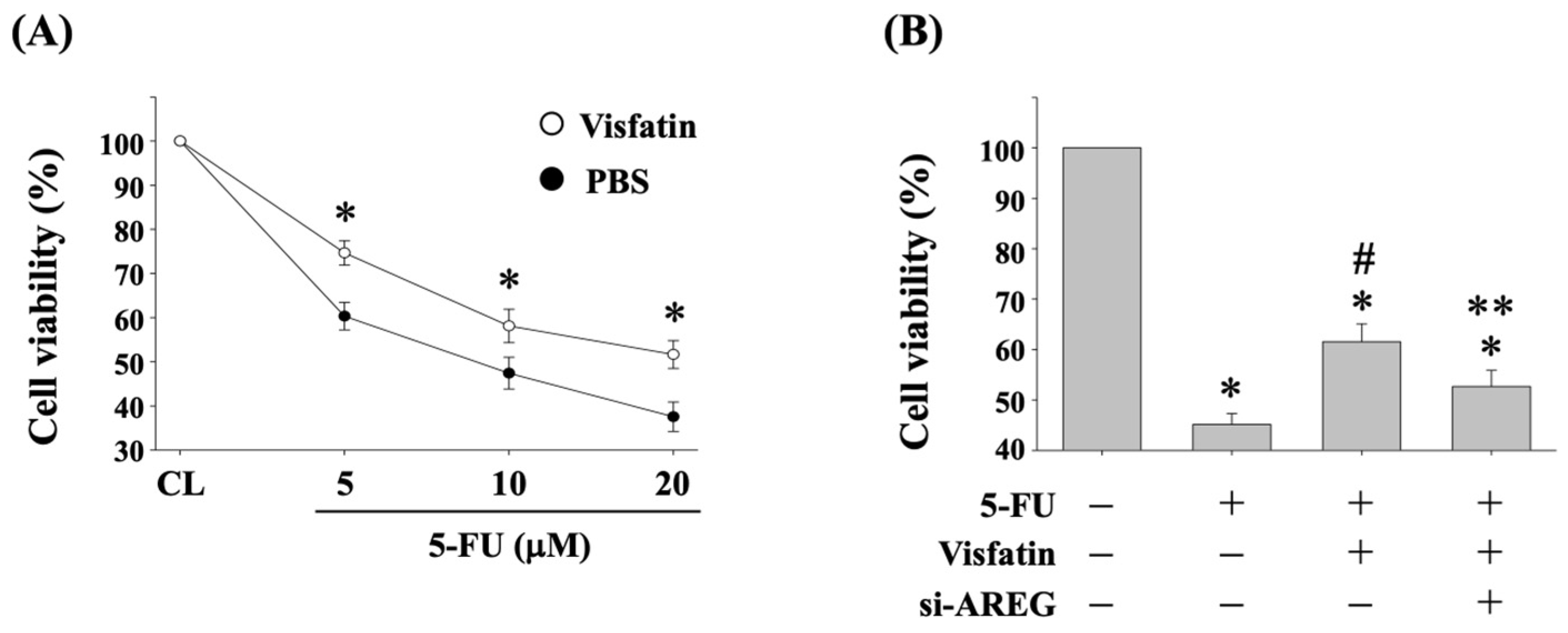
3.2. Visfatin-Increased AREG Levels Attenuate the Cytotoxicity of HCT-116 CRC Cells in Response to 5-FU
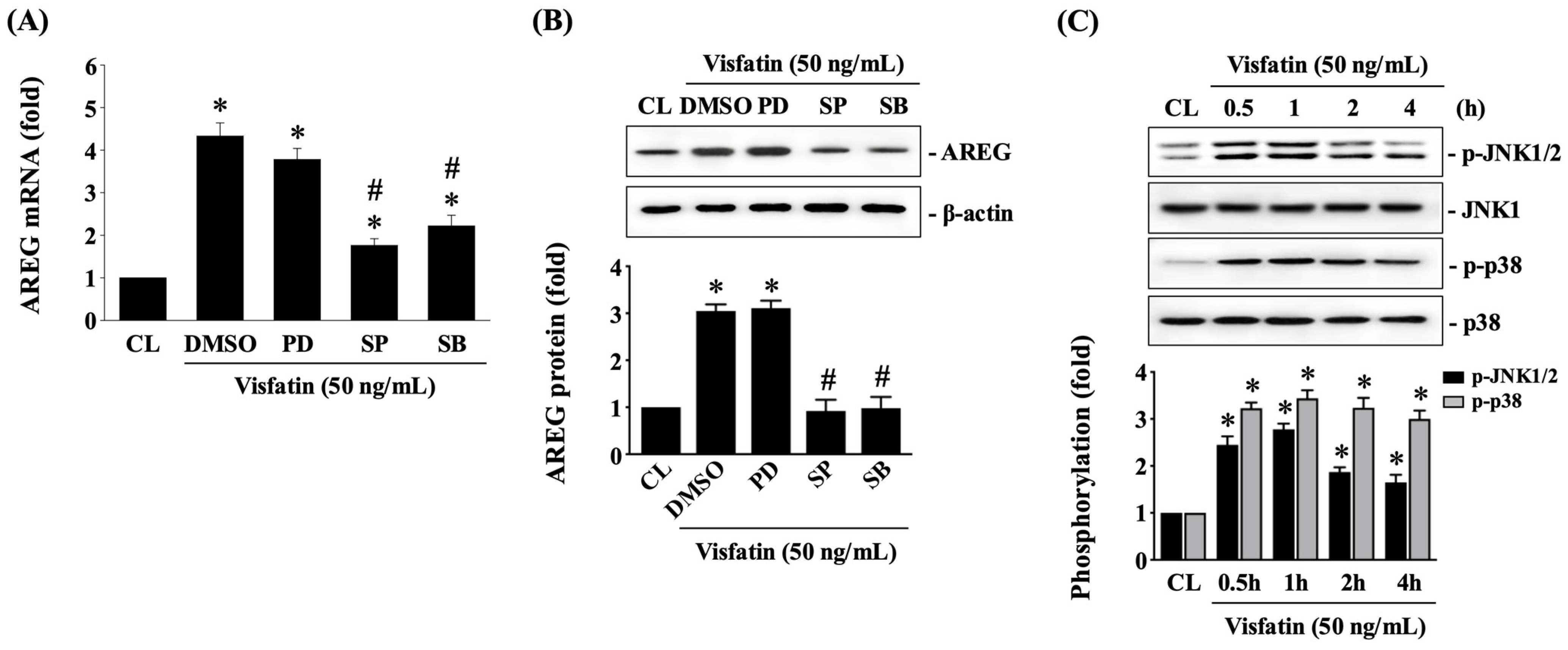
3.3. JNK1/2 and p38 Signaling Regulate AREG Upregulation in HCT-116 CRC Cells under Visfatin Stimulation
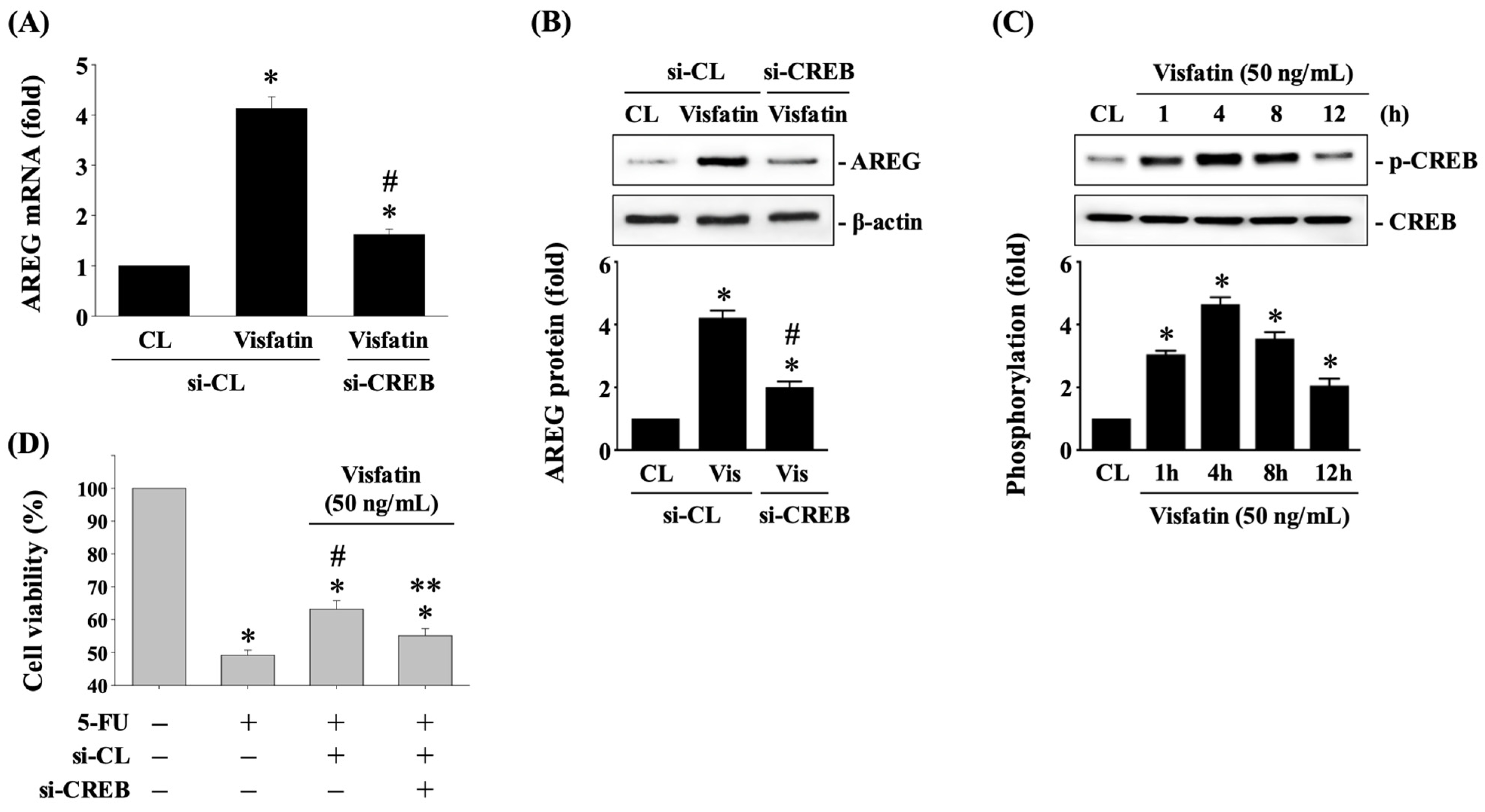
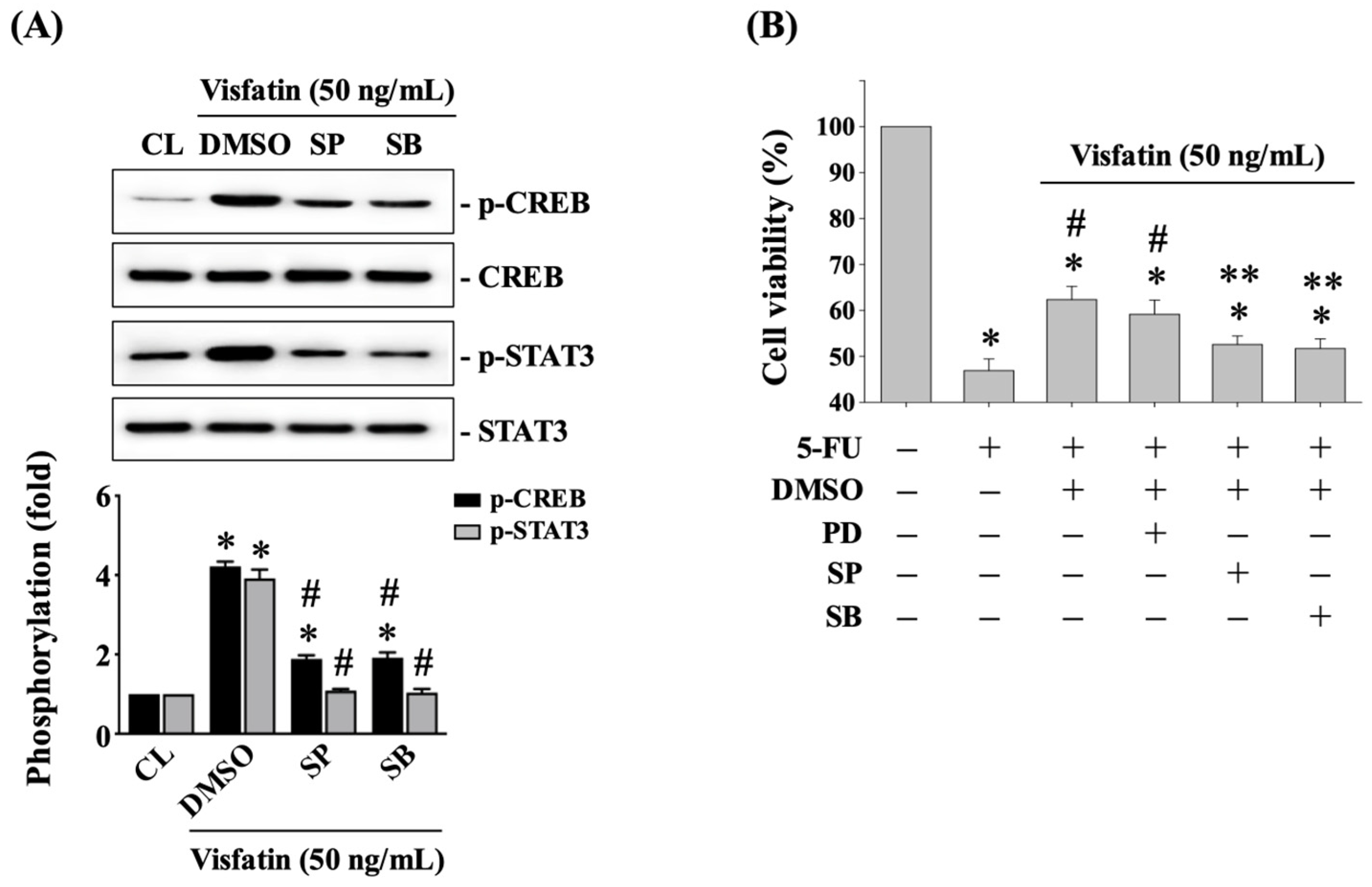
3.4. CREB Affects AREG Upregulation and Subsequent 5-FU-Initiated Cytotoxicity in Visfatin-Stimulated HCT-116 CRC Cells
3.5. STAT3 Affects AREG Upregulation and Subsequent 5-FU-Initiated Cytotoxicity in Visfatin-Stimulated HCT-116 CRC Cells
3.6. JNK1/2 and p38 Signaling Affects CREB and STAT3 Phosphorylation and Subsequent 5-FU-Initiated Cytotoxicity in Visfatin-Stimulated HCT-116 CRC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.J.; Chiu, H.-M.; Jung, K.-W.; Jun, J.K.; Sekiguchi, M.; Matsuda, T.; Kyaw, M.H. Increasing Trend in Young-Onset Colorectal Cancer in Asia: More Cancers in Men and More Rectal Cancers. Am. J. Gastroenterol. 2019, 114, 322–329. [Google Scholar] [CrossRef]
- Parmar, S.; Easwaran, H. Genetic and epigenetic dependencies in colorectal cancer development. Gastroenterol. Rep. 2022, 10, goac035. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Biller, L.H.; Schrag, D. Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, X.; Chen, G.; Du, J. Drug Resistance in Colorectal Cancer: From Mechanism to Clinic. Cancers 2022, 14, 2928. [Google Scholar] [CrossRef]
- Ashique, S.; Bhowmick, M.; Pal, R.; Khatoon, H.; Kumar, P.; Sharma, H.; Garg, A.; Kumar, S.; Das, U. Multi drug resistance in Colorectal Cancer- approaches to overcome, advancements and future success. Adv. Cancer Biol.—Metastasis 2024, 10, 100114. [Google Scholar] [CrossRef]
- Hu, T.; Li, Z.; Gao, C.Y.; Cho, C.H. Mechanisms of drug resistance in colon cancer and its therapeutic strategies. World J. Gastroenterol. 2016, 22, 6876–6889. [Google Scholar] [CrossRef]
- Berasain, C.; Avila, M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014, 28, 31–41. [Google Scholar] [CrossRef]
- Zaiss, D.M.W.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging Functions of Amphiregulin in Orchestrating Immunity, Inflammation, and Tissue Repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef]
- Singh, S.S.; Chauhan, S.B.; Kumar, A.; Kumar, S.; Engwerda, C.R.; Sundar, S.; Kumar, R. Amphiregulin in cellular physiology, health, and disease: Potential use as a biomarker and therapeutic target. J. Cell. Physiol. 2022, 237, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chiao, P.; Sun, Y. Amphiregulin in Cancer: New Insights for Translational Medicine. Trends Cancer 2016, 2, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-A.; Park, H.; Kim, K.-J.; Sung, J.H.; Nam, M.; Lee, J.H.; Jung, E.H.; Suh, K.J.; Lee, J.Y.; Kim, S.H.; et al. Amphiregulin can predict treatment resistance to palliative first-line cetuximab plus FOLFIRI chemotherapy in patients with RAS wild-type metastatic colorectal cancer. Sci. Rep. 2021, 11, 23803. [Google Scholar] [CrossRef] [PubMed]
- Avgerinos, K.I.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Obesity and cancer risk: Emerging biological mechanisms and perspectives. Metabolism 2019, 92, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Akrida, I.; Papadaki, H. Adipokines and epithelial-mesenchymal transition (EMT) in cancer. Mol. Cell. Biochem. 2023, 478, 2419–2433. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zhao, X.; Chen, M.; Ji, H.; Zhang, Q.; Chen, R.; Wang, Y. Plasma adiponectin, visfatin, leptin, and resistin levels and the onset of colonic polyps in patients with prediabetes. BMC Endocr. Disord. 2020, 20, 63. [Google Scholar] [CrossRef]
- Chen, M.; Wang, Y.; Li, Y.; Zhao, L.; Ye, S.; Wang, S.; Yu, C.; Xie, H. Association of plasma visfatin with risk of colorectal cancer: An observational study of Chinese patients. Asia Pac. J. Clin. Oncol. 2016, 12, e65–e74. [Google Scholar] [CrossRef]
- Lin, T.C. The role of visfatin in cancer proliferation, angiogenesis, metastasis, drug resistance and clinical prognosis. Cancer Manag. Res. 2019, 11, 3481–3491. [Google Scholar] [CrossRef]
- Ji, C.; Cong, R.; Wang, Y.; Wang, Y.; Zhang, Q.; Zhou, X.; Xing, Q.; Song, N. Relationship between NAMPT/PBEF/visfatin and prognosis of patients with malignant tumors: A systematic review and meta-analysis. Ann. Transl. Med. 2019, 7, 785. [Google Scholar] [CrossRef]
- Wu, K.L.; Lee, K.C.; Yen, C.K.; Chen, C.N.; Chang, S.F.; Huang, W.S. Visfatin and Resveratrol Differentially Regulate the Expression of Thymidylate Synthase to Control the Sensitivity of Human Colorectal Cancer Cells to Capecitabine Cytotoxicity. Life 2021, 11, 1371. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, W.; Yu, T.; Cui, X.; Zhou, Z.; Yang, H.; Yu, Y.; Bilotta, A.J.; Yao, S.; Xu, J.; et al. Th17 Cell-Derived Amphiregulin Promotes Colitis-Associated Intestinal Fibrosis Through Activation of mTOR and MEK in Intestinal Myofibroblasts. Gastroenterology 2023, 164, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Xiang, H.; Zhang, W. Review of various NAMPT inhibitors for the treatment of cancer. Front. Pharmacol. 2022, 13, 970553. [Google Scholar] [CrossRef] [PubMed]
- Travelli, C.; Colombo, G.; Aliotta, M.; Fagiani, F.; Fava, N.; De Sanctis, R.; Grolla, A.A.; Garcia, J.G.N.; Clemente, N.; Portararo, P.; et al. Extracellular nicotinamide phosphoribosyltransferase (eNAMPT) neutralization counteracts T cell immune evasion in breast cancer. J. Immunother. Cancer 2023, 11, e007010. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-H.; Hsu, H.-C.; You, J.-F.; Lai, C.-C.; Tsou, Y.-K.; Hsu, C.-L.; Fann, C.S.J.; Chien, R.-N.; Chang, M.-L. Extracellular Nicotinamide Phosphoribosyltransferase as a Surrogate Marker of Prominent Malignant Potential in Colonic Polyps: A 2-Year Prospective Study. Cancers 2023, 15, 1702. [Google Scholar] [CrossRef]
- Yu, J.; Li, X.; Zhong, C.; Li, D.; Zhai, X.; Hu, W.; Guo, C.; Yuan, Y.; Zheng, S. High-throughput proteomics integrated with gene microarray for discovery of colorectal cancer potential biomarkers. Oncotarget 2016, 7, 75279–75292. [Google Scholar] [CrossRef]
- Teplan, V.; Senolt, L.; Hulejova, H.; Teplan, V.; Stollova, M.; Gurlich, R. Early changes in serum visfatin after abdominal surgery: A new pro-inflammatory marker in diagnosis? Biomed. Pap. Med. Fac. Palacky Univ. Olomouc Czech Repub. 2015, 159, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, K.; Misa, I.B.; Diakowska, D.; Kapturkiewicz, B.; Gamian, A.; Krzystek-Korpacka, M. Nampt/PBEF/visfatin upregulation in colorectal tumors, mirrored in normal tissue and whole blood of colorectal cancer patients, is associated with metastasis, hypoxia, IL1β, and anemia. Biomed. Res. Int. 2015, 2015, 523930. [Google Scholar] [CrossRef]
- Zhao, Q.; Long, Y.; Cheng, W.; Huang, Y.; Li, J.; Li, Y.; Li, X.; Guo, X.; Li, Y.; Li, G.; et al. Visfatin inhibits colon cancer cell apoptosis and decreases chemosensitivity to 5-FU by promoting the SDF-1/CXCR4/Akt axis. Int. J. Oncol. 2022, 60, 75. [Google Scholar] [CrossRef]
- Lucena-Cacace, A.; Otero-Albiol, D.; Jiménez-García, M.P.; Muñoz-Galvan, S.; Carnero, A. NAMPT Is a Potent Oncogene in Colon Cancer Progression that Modulates Cancer Stem Cell Properties and Resistance to Therapy through Sirt1 and PARP. Clin. Cancer Res. 2018, 24, 1202–1215. [Google Scholar] [CrossRef]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.L.; Hurbin, A. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta 2011, 1816, 119–131. [Google Scholar] [CrossRef] [PubMed]
- HHong, C.; Sun, E.; Choi, J.; Kim, D.; Kim, J.; Ryu, K.; Shim, H.; Hwang, J.; Bae, W.; Kim, H.; et al. Fibroblast growth factor receptor 4 increases epidermal growth factor receptor (EGFR) signaling by inducing amphiregulin expression and attenuates response to EGFR inhibitors in colon cancer. Cancer Sci. 2020, 111, 3268–3278. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, S.; Wang, B.; Wu, Y.; Chen, Z.; Lv, M.; Lin, Y.; Yang, J. Potential biomarkers for anti-EGFR therapy in metastatic colorectal cancer. Tumour Biol. 2016, 37, 11645–11655. [Google Scholar] [CrossRef] [PubMed]
- O’reilly, S.M.; Leonard, M.O.; Kieran, N.; Comerford, K.M.; Cummins, E.; Pouliot, M.; Lee, S.B.; Taylor, C.T. Hypoxia induces epithelial amphiregulin gene expression in a CREB-dependent manner. Am. J. Physiol.-Cell Physiol. 2006, 290, C592–C600. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Operario, D.J.; Georas, S.N.; Mosmann, T.R. The acute environment, rather than T cell subset pre-commitment, regulates expression of the human T cell cytokine amphiregulin. PLoS ONE 2012, 7, e39072. [Google Scholar] [CrossRef]
- Du, B.; Altorki, N.K.; Kopelovich, L.; Subbaramaiah, K.; Dannenberg, A.J. Tobacco smoke stimulates the transcription of amphiregulin in human oral epithelial cells: Evidence of a cyclic AMP-responsive element binding protein-dependent mechanism. Cancer Res. 2005, 65, 5982–5988. [Google Scholar] [CrossRef]
- Johansson, C.C.; Yndestad, A.; Enserink, J.M.; Ree, A.H.; Aukrust, P.; Taskén, K. The epidermal growth factor-like growth factor amphiregulin is strongly induced by the adenosine 3′,5′-monophosphate pathway in various cell types. Endocrinology 2004, 145, 5177–5184. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Lu, Z.; Fang, Z.; Guo, Y.; Liu, X.; Chen, S. Cisplatin resistance of NSCLC cells involves upregulation of visfatin through activation of its transcription and stabilization of mRNA. Chem. Biol. Interact. 2022, 351, 109705. [Google Scholar] [CrossRef]
- Tahergorabi, Z.; Lotfi, H.; Rezaei, M.; Aftabi, M.; Moodi, M. Crosstalk between obesity and cancer: A role for adipokines. Arch. Physiol. Biochem. 2024, 130, 155–168. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, W.-S.; Wu, K.-L.; Chen, C.-N.; Chang, S.-F.; Lee, D.-Y.; Lee, K.-C. Amphiregulin Upregulation in Visfatin-Stimulated Colorectal Cancer Cells Reduces Sensitivity to 5-Fluororacil Cytotoxicity. Biology 2024, 13, 821. https://doi.org/10.3390/biology13100821
Huang W-S, Wu K-L, Chen C-N, Chang S-F, Lee D-Y, Lee K-C. Amphiregulin Upregulation in Visfatin-Stimulated Colorectal Cancer Cells Reduces Sensitivity to 5-Fluororacil Cytotoxicity. Biology. 2024; 13(10):821. https://doi.org/10.3390/biology13100821
Chicago/Turabian StyleHuang, Wen-Shih, Kuen-Lin Wu, Cheng-Nan Chen, Shun-Fu Chang, Ding-Yu Lee, and Ko-Chao Lee. 2024. "Amphiregulin Upregulation in Visfatin-Stimulated Colorectal Cancer Cells Reduces Sensitivity to 5-Fluororacil Cytotoxicity" Biology 13, no. 10: 821. https://doi.org/10.3390/biology13100821
APA StyleHuang, W.-S., Wu, K.-L., Chen, C.-N., Chang, S.-F., Lee, D.-Y., & Lee, K.-C. (2024). Amphiregulin Upregulation in Visfatin-Stimulated Colorectal Cancer Cells Reduces Sensitivity to 5-Fluororacil Cytotoxicity. Biology, 13(10), 821. https://doi.org/10.3390/biology13100821