
Transcriptomic and Metabolomic Analyses Reveal the Response Mechanism of Ophiopogon japonicus to Waterlogging Stress

Abstract
:Simple Summary
Abstract
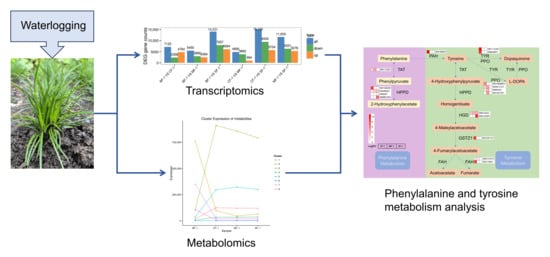
1. Introduction
2. Materials and Methods
2.1. Plant Material and Growing Conditions
2.2. Transcriptome Analysis
2.3. Metabolite Identification
2.4. Real-Time Quantitative PCR Validation
2.5. Statistical Analysis
3. Results
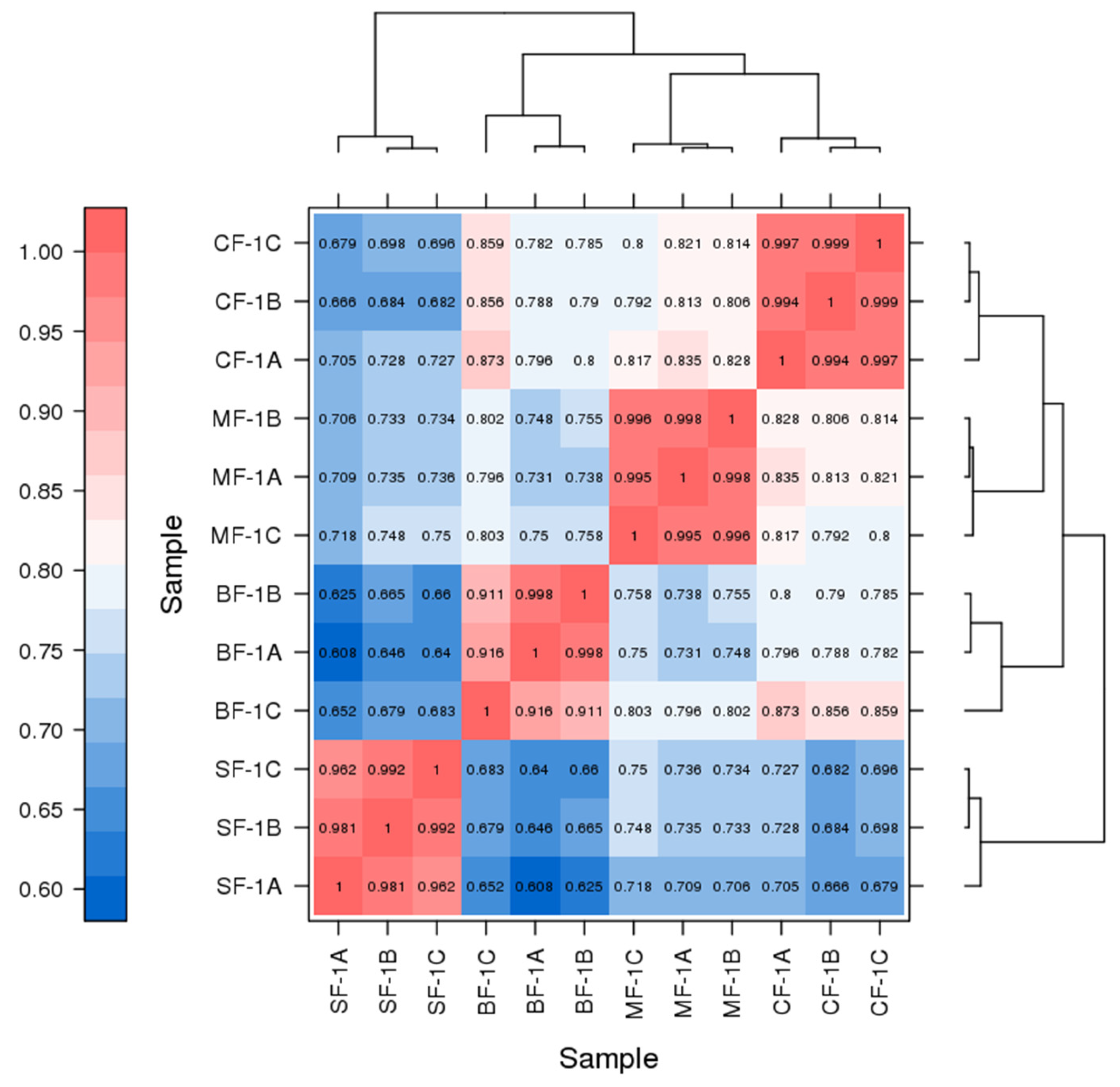
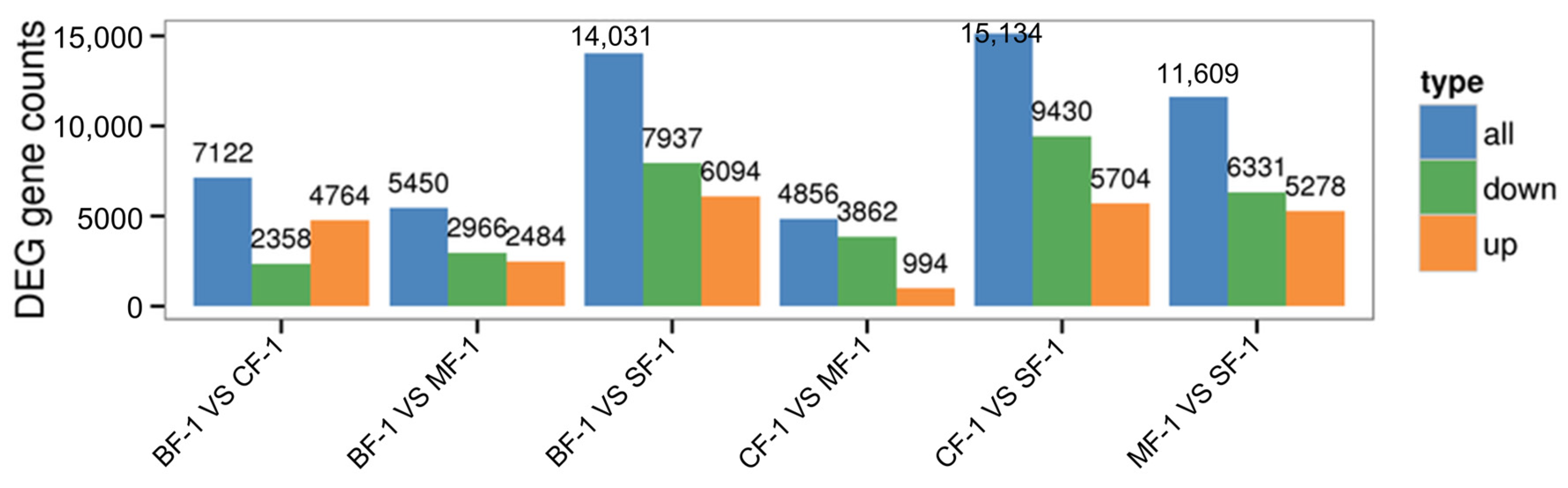
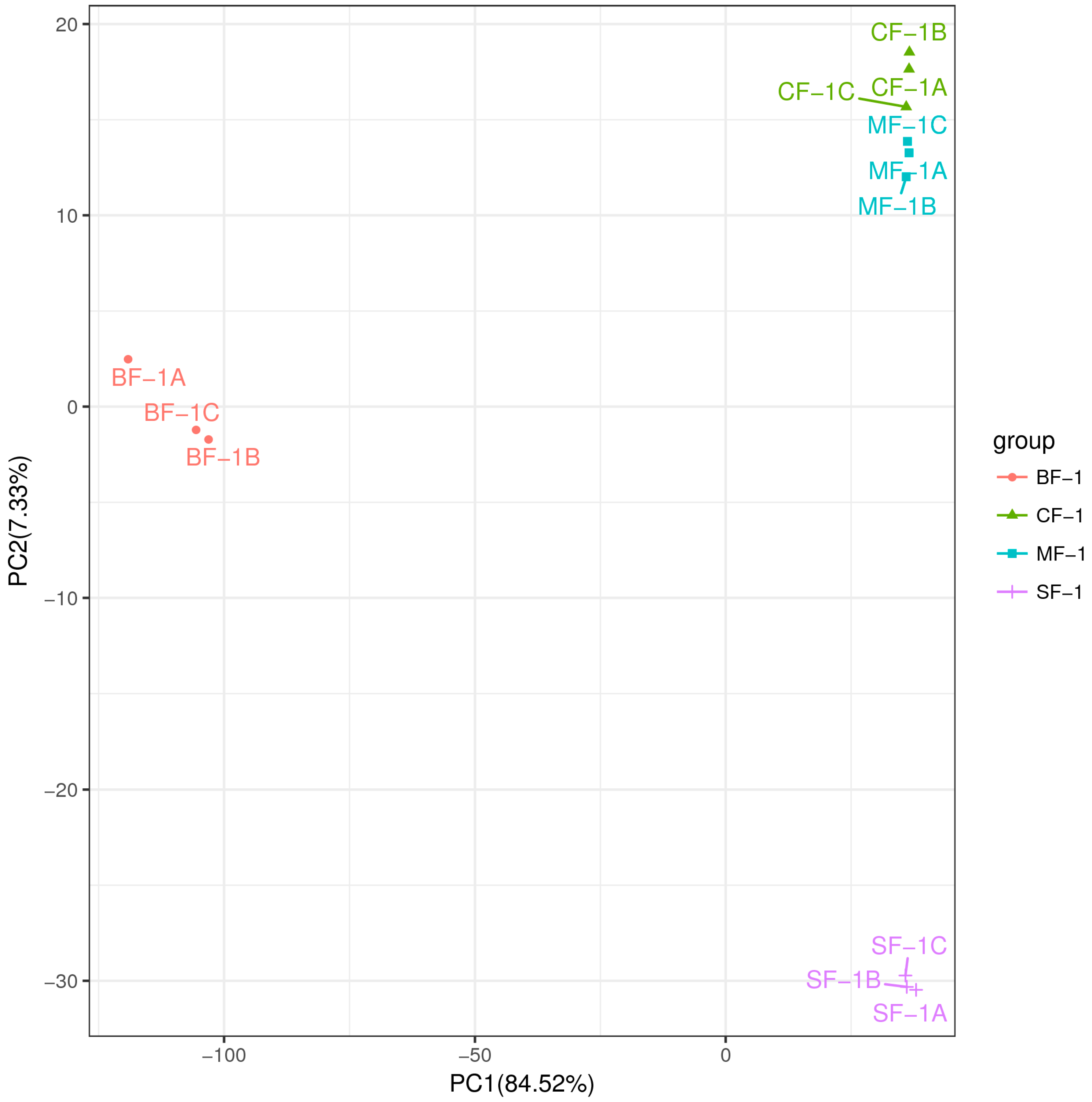
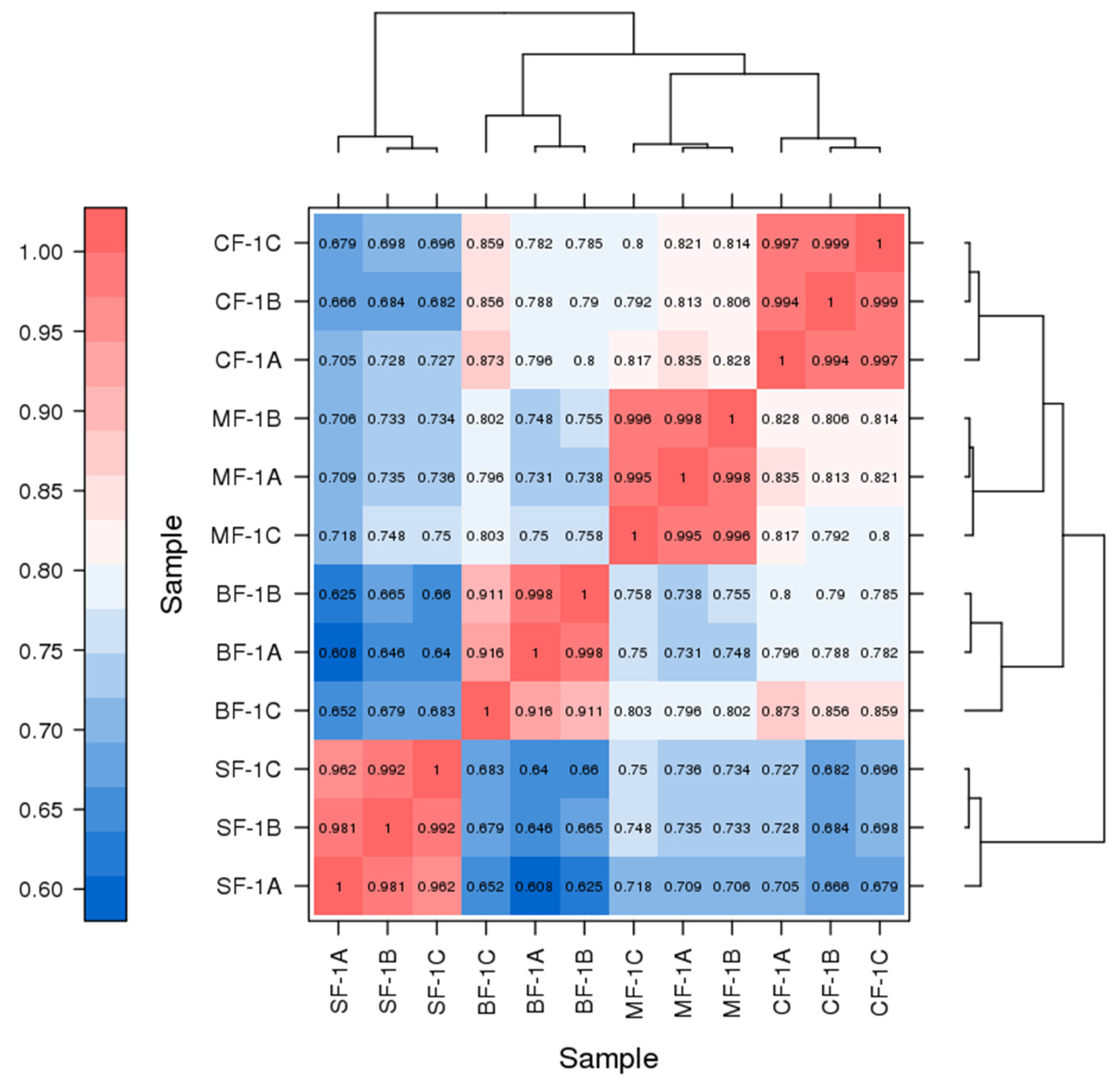
3.1. Analysis of Transcriptome Results
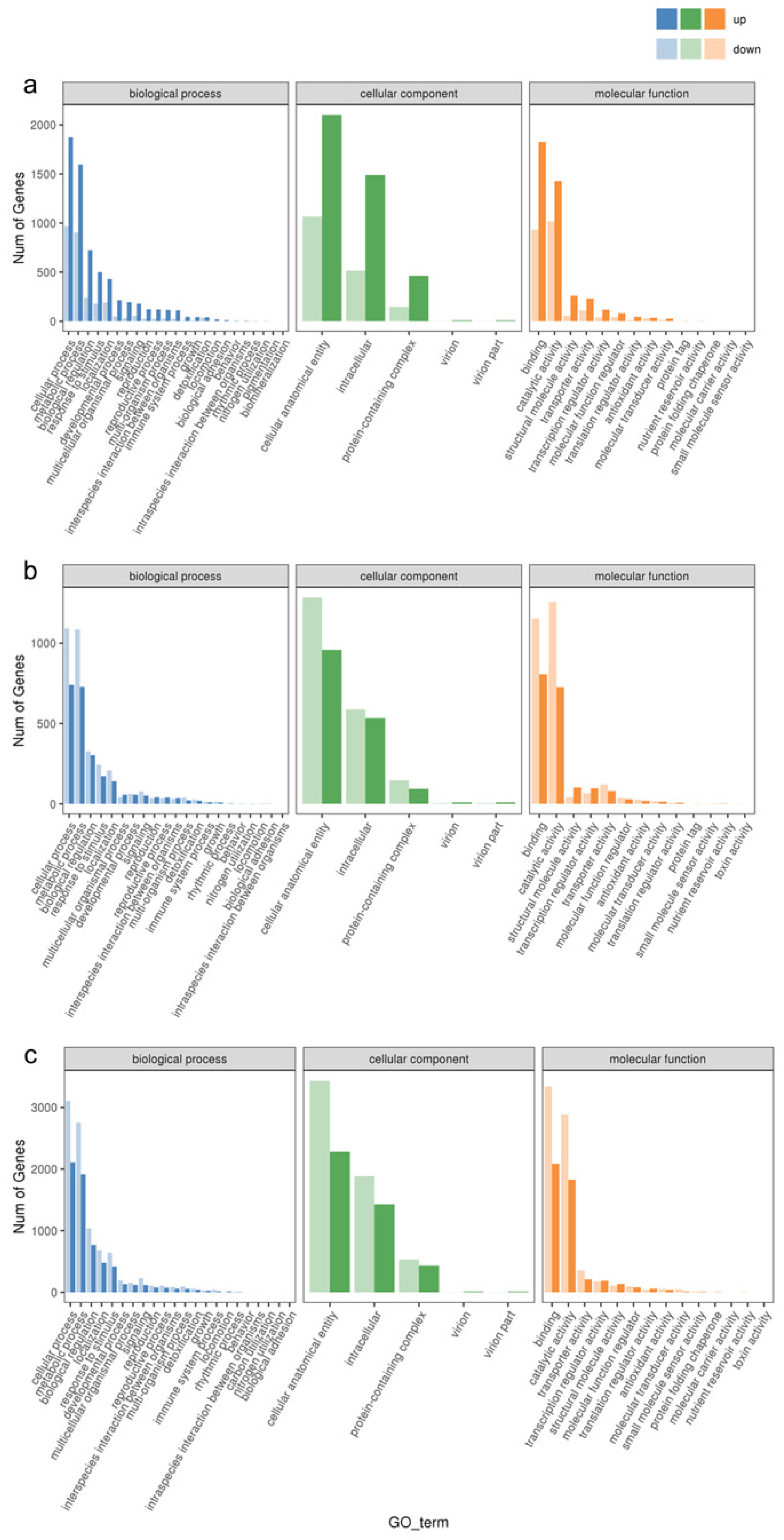
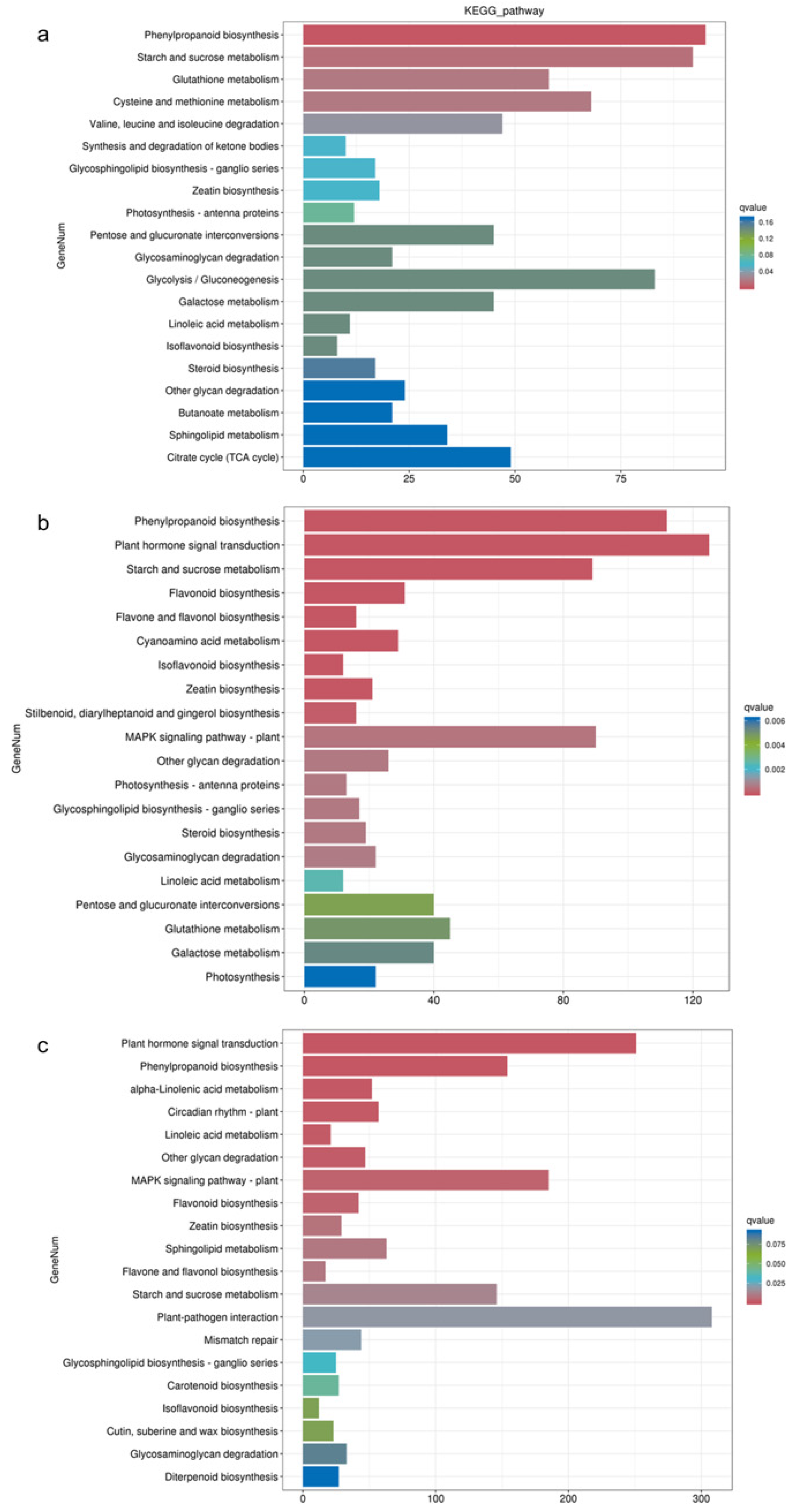
3.2. Differential Gene Function Analysis
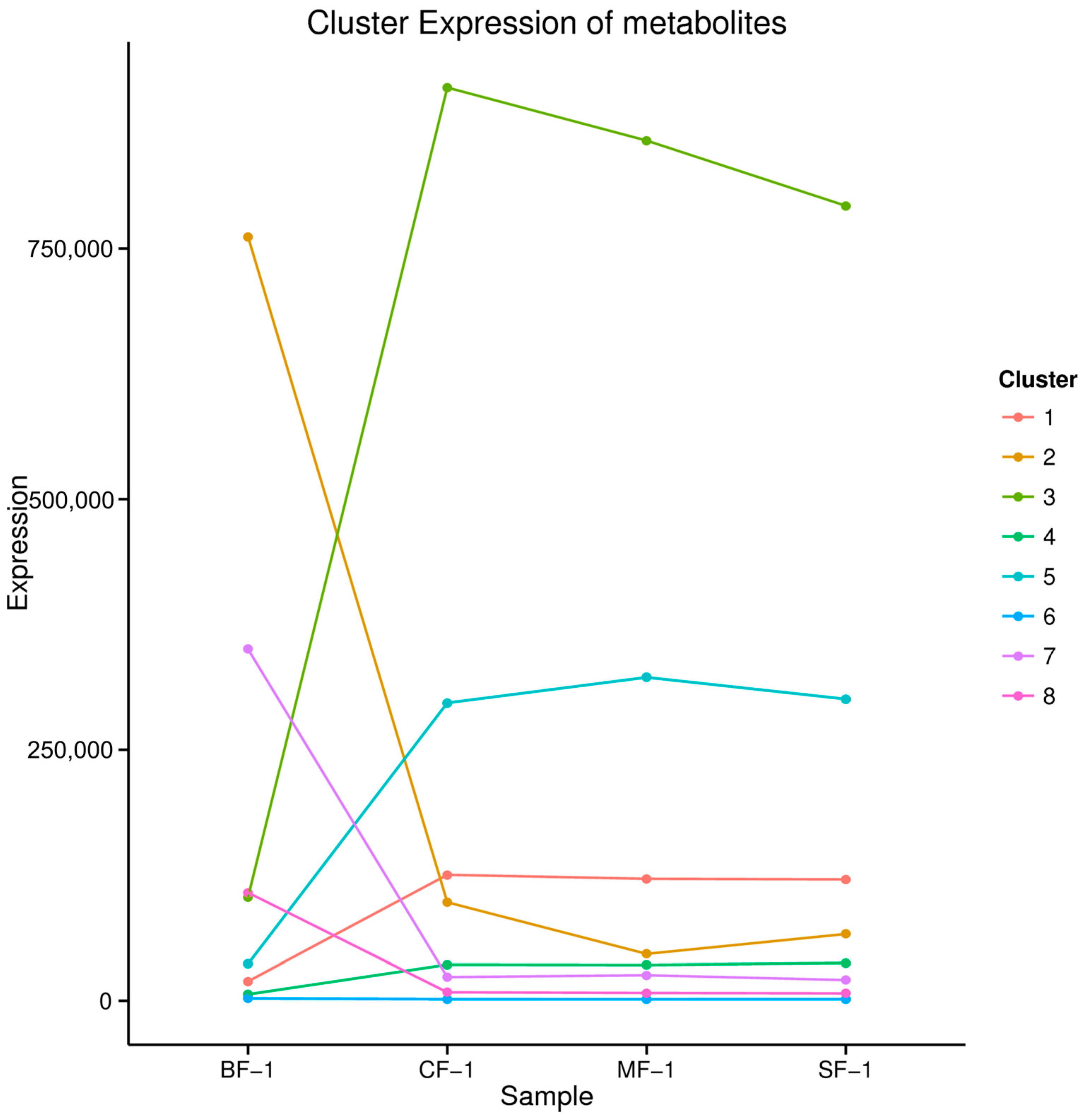
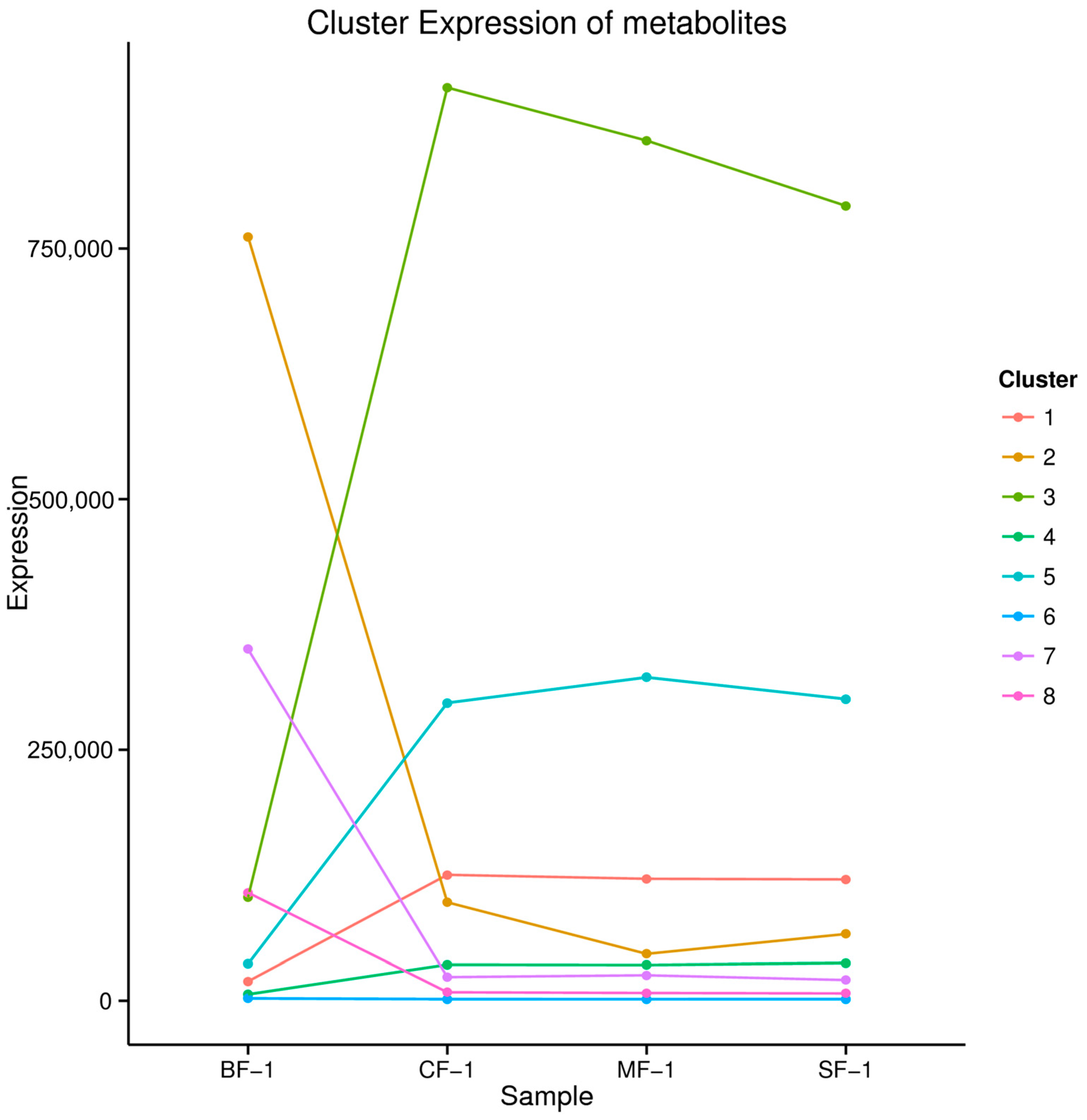
3.3. Metabolomic Analysis
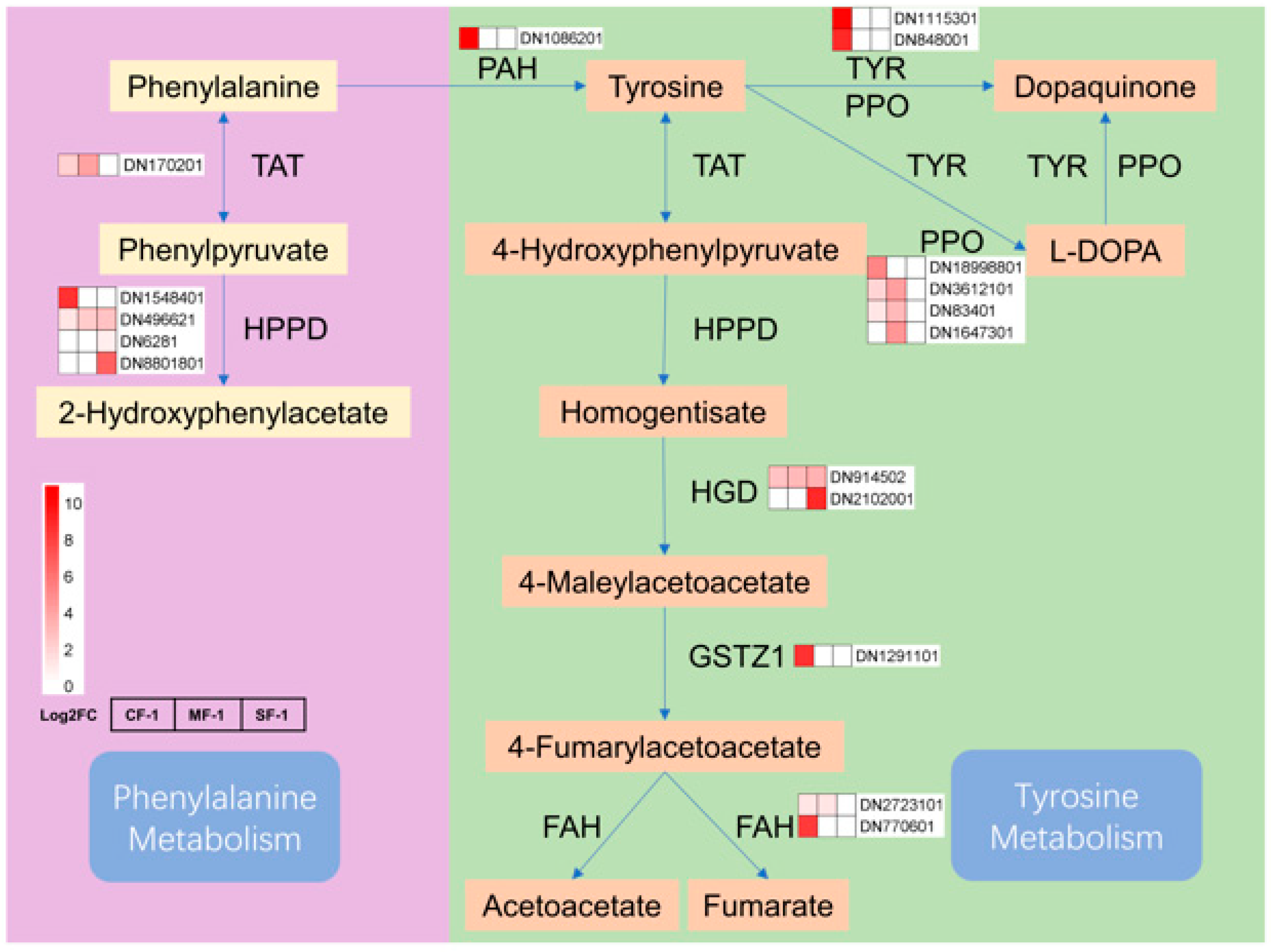
3.4. The Material Basis of CMD Response to Waterlogging Stress
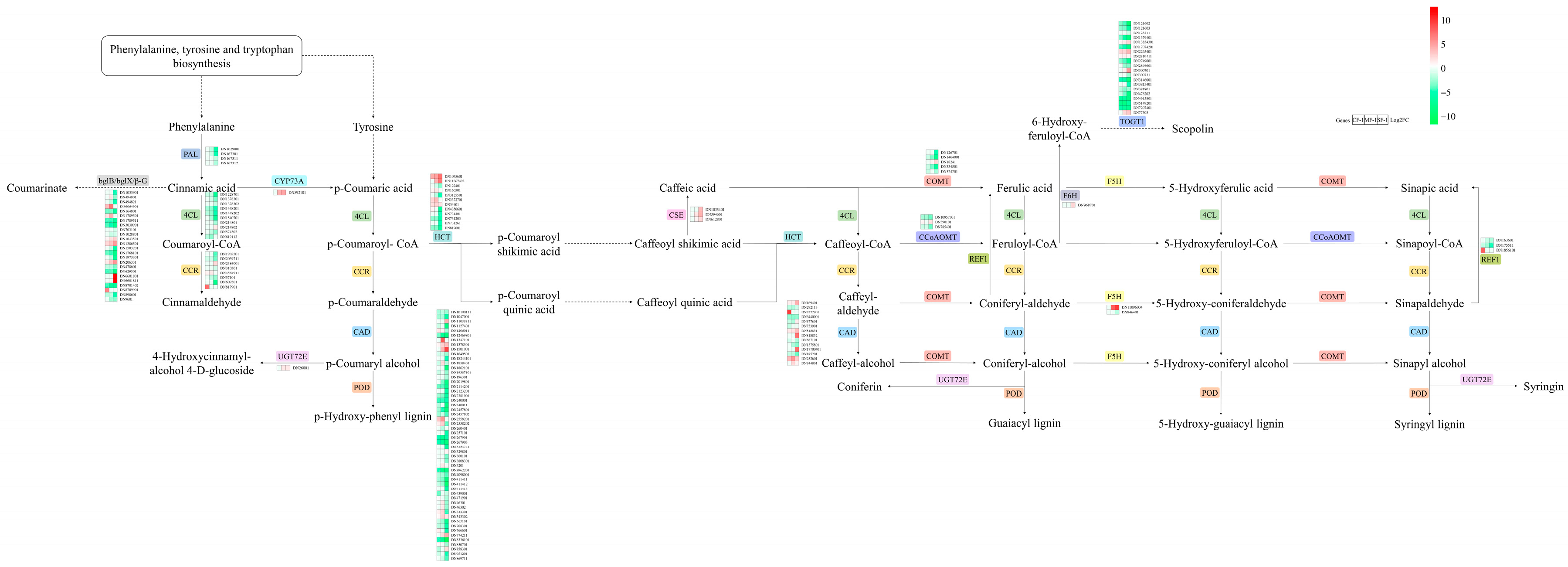
3.5. Key Pathways of CMD in Response to Waterlogging Stresses
3.6. Key TFs of CMD in Response to Waterlogging Stresses
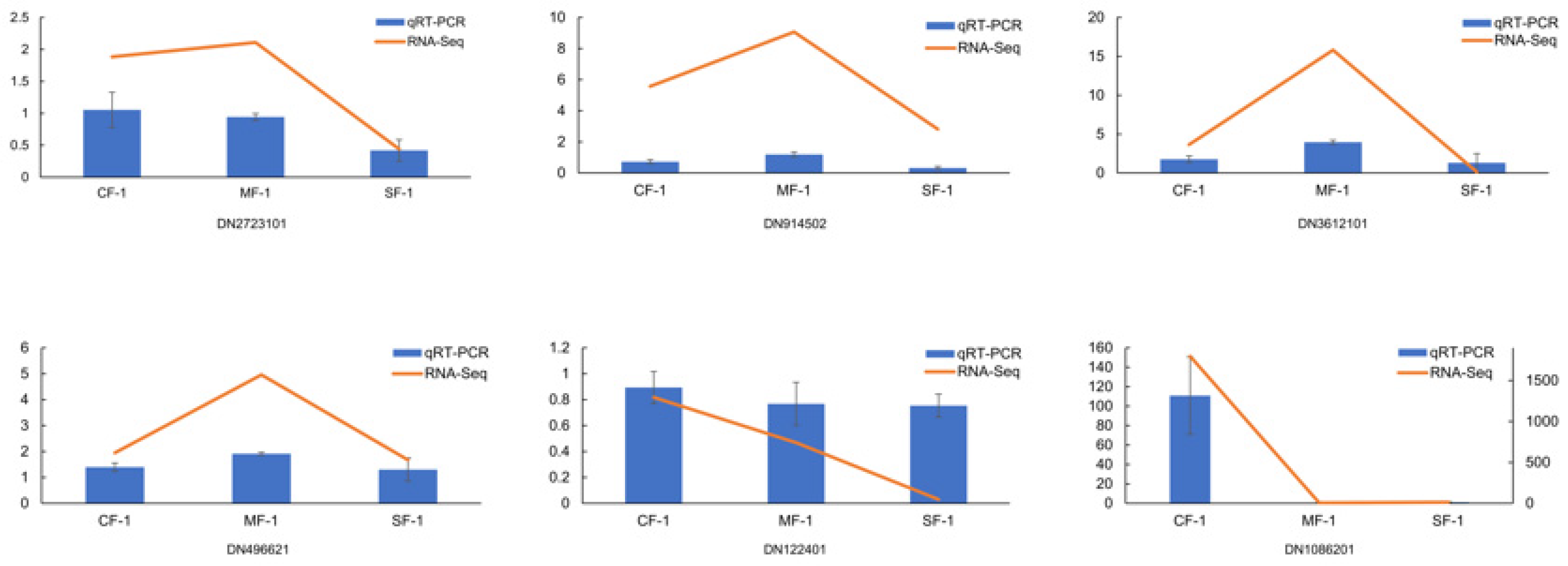
3.7. Validation of qRT-PCR
4. Discussion
4.1. Impact of Waterlogging on the CMD Transcriptome and Metabolome
4.2. Significantly Adjusted Biosynthesis Responses in Waterlogged CMD
4.3. Differentially Expressed TF Responses in Waterlogged CMD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.-F.; Wang, L.-L.; Chen, L.-C.; Liu, T.-B.; Sha, R.-Y.; Mao, J.-W. Enrichment and Separation of Steroidal Saponins from the Fibrous Roots of Ophiopogon japonicus Using Macroporous Adsorption Resins. RSC Adv. 2019, 9, 6689–6698. [Google Scholar] [CrossRef]
- Zou, X.; Hu, C.; Zeng, L.; Cheng, Y.; Xu, M.; Zhang, X. A Comparison of Screening Methods to Identify Waterlogging Tolerance in the Field in Brassica napus L. during Plant Ontogeny. PLoS ONE 2014, 9, e89731. [Google Scholar] [CrossRef]
- Abbas, F.; Guo, S.; Zhou, Y.; Wu, J.; Amanullah, S.; Wang, H.-C.; Shen, J. Metabolome and Transcriptome Analysis of Terpene Synthase Genes and Their Putative Role in Floral Aroma Production in Litchi chinensis. Physiol. Plant 2022, 174, e13796. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Li, B.; Tian, H.; Li, X.; Ren, H.; Zhou, Q. Metabolome and Transcriptome Analysis Predicts Metabolism of Violet-Red Color Change in Lilium Bulbs. J. Sci. Food Agric. 2022, 102, 2903–2915. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhang, X.; Wang, Q.; Guo, Y.; Xie, H.; Li, L.; Zhang, P.; Liu, J.; Qin, P. Metabolome and Transcriptome Profiles in Quinoa Seedlings in Response to Potassium Supply. BMC Plant Biol. 2022, 22, 604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yan, Y.; Zhou, W.-H.; Feng, R.-Z.; Shuai, Y.-K.; Yang, L.; Liu, M.-J.; He, X.-Y.; Wei, Q. Transcriptome and Metabolome Reveal the Accumulation of Secondary Metabolites in Different Varieties of Cinnamomum longepaniculatum. BMC Plant Biol. 2022, 22, 243. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhan, X.; Liu, Y.; Huang, Z.; Deng, X.; Zheng, Z.; Xu, M. Integrated Transcriptome and Metabolome Analysis Reveals Phenylpropanoid Biosynthesis and Phytohormone Signaling Contribute to “Candidatus Liberibacter Asiaticus” Accumulation in Citrus Fruit Piths (Fluffy Albedo). Int. J. Mol. Sci. 2022, 23, 15648. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Xu, Z.; Huang, W.; Li, D.; Zeng, Q.; Chen, L.; Li, B.; Zhang, E. Integrated Metabolome and Transcriptome Analysis Reveals Salicylic Acid and Flavonoid Pathways’ Key Roles in Cabbage’s Defense Responses to Xanthomonas campestris Pv. campestris. Front. Plant Sci. 2022, 13, 1005764. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, T.; Gao, M.; Ye, M.; Lin, M.; Wu, D.; Guo, J.; Guan, W.; Wang, J.; Yang, K.; et al. Transcriptome and Metabolome Profiling Reveal the Resistance Mechanisms of Rice against Brown Planthopper. Int. J. Mol. Sci. 2022, 23, 4083. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Q.; Tan, Y.; Shuang, S.; Dai, R.; Jiang, X.; Temuer, B. Combined Transcriptome and Metabolome Analysis of Alfalfa Response to Thrips Infection. Genes 2021, 12, 1967. [Google Scholar] [CrossRef]
- Cui, J.; Li, J.; Dai, C.; Li, L. Transcriptome and Metabolome Analyses Revealed the Response Mechanism of Sugar Beet to Salt Stress of Different Durations. Int. J. Mol. Sci. 2022, 23, 9599. [Google Scholar] [CrossRef]
- Dwivedi, A.K.; Singh, V.; Anwar, K.; Pareek, A.; Jain, M. Integrated Transcriptome, Proteome and Metabolome Analyses Revealed Secondary Metabolites and Auxiliary Carbohydrate Metabolism Augmenting Drought Tolerance in Rice. Plant Physiol. Biochem. 2023, 201, 107849. [Google Scholar] [CrossRef]
- Jo, J.; Lee, J.; Ahn, Y.; Hwang, Y.S.; Park, J.; Lee, J.; Choi, J. Metabolome and Transcriptome Analyses of Plants Grown in Naturally Attenuated Soil after Hydrogen Fluoride Exposure. J. Hazard. Mater. 2022, 437, 129323. [Google Scholar] [CrossRef] [PubMed]
- Owusu, A.G.; Lv, Y.-P.; Liu, M.; Wu, Y.; Li, C.-L.; Guo, N.; Li, D.-H.; Gao, J.-S. Transcriptomic and Metabolomic Analyses Reveal the Potential Mechanism of Waterlogging Resistance in Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2023, 14, 1088537. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-H.; Leong, F.; Gao, S.-J.; Chen, X.; Lu, J.-J.; Lin, L.-G.; Wang, Y.; Chen, X.-J. Comparison of Ophiopogon japonicus and Liriope Spicata Var. Prolifera from Different Origins Based on Multi-Component Quantification and Anticancer Activity. Molecules 2023, 28, 1045. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-Y.; Liu, Y.-L.; Shen, Y.; Zhang, Q.-Y.; Liu, S.; Ren, Q.-R.; Qin, L.-P.; Sun, Y.-Q. Phylogeography of Cultivated and Wild Ophiopogon japonicus Based on Chloroplast DNA: Exploration of the Origin and Sustainable Cultivation. BMC Plant Biol. 2023, 23, 242. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, B.; Peng, X.; Li, S.; Zhao, J. Quality Evaluation of Ophiopogon japonicus from Two Authentic Geographical Origins in China Based on Physicochemical and Pharmacological Properties of Their Polysaccharides. Biomolecules 2022, 12, 1491. [Google Scholar] [CrossRef]
- Alam, I.; Lee, D.-G.; Kim, K.-H.; Park, C.-H.; Sharmin, S.A.; Lee, H.; Oh, K.-W.; Yun, B.-W.; Lee, B.-H. Proteome Analysis of Soybean Roots under Waterlogging Stress at an Early Vegetative Stage. J. Biosci. 2010, 35, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, F.; Lu, C.; Wang, F.; Wen, Y.; Liu, Y.; Gao, S.; Qi, W.; Li, X.; Yang, J. First Report of Tobacco Mild Green Mosaic Virus Infecting Rehmannia glutinosa in China. Plant Dis. 2022, 106, 3004. [Google Scholar] [CrossRef]
- Panailidou, P.; Lotos, L.; Sassalou, C.-L.; Gagiano, E.; Pietersen, G.; Katis, N.; Maliogka, V.I. First Report of Grapevine Virus H (GVH) in Grapevine in Greece. Plant Dis. 2021, 105, 2738. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Ni, L.; Carballar-Lejarazú, R.; Zou, X.; Sun, W.; Wu, L.; Yuan, X.; Mao, Y.; Huang, W.; Zou, S. Comparative Transcriptome among Euscaphis konishii Hayata Tissues and Analysis of Genes Involved in Flavonoid Biosynthesis and Accumulation. BMC Genom. 2019, 20, 24. [Google Scholar] [CrossRef]
- Chen, C.; Xu, L.; Xia, W.; Qi, C.; Liu, J.; Hua, B.; Zhang, Z.; Qi, X.; Miao, M. Metabolomics and Transcription Profiling of Pumpkin Fruit Reveals Enhanced Bioactive Flavonoids and Coumarins in Giant Pumpkin (Cucurbita maxima). J. Agric. Food Chem. 2023, 71, 10459–10469. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Le Provost, G.; Lesur, I.; Lalanne, C.; Da Silva, C.; Labadie, K.; Aury, J.M.; Leple, J.C.; Plomion, C. Implication of the Suberin Pathway in Adaptation to Waterlogging and Hypertrophied Lenticels Formation in Pedunculate Oak (Quercus robur L.). Tree Physiol. 2016, 36, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Long, Y.; Yin, X.; Wang, W.; Li, W.; Chen, T.; Chen, J.; Chen, X.; Wang, B.; Ma, J. Metabolome and Transcriptome Analysis of Sulfur-Induced Kiwifruit Stem Laccase Gene Involved in Syringyl Lignin Synthesis against Bacterial Canker. J. Agric. Food Chem. 2023, 71, 13566–13576. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Zhou, F.; Xu, D.; Xiong, S.; Chen, C.; Liu, C.; Jiang, A. Transcriptomic and Metabolomic Profiling Reveal the Mechanism Underlying the Inhibition of Wound Healing by Ascorbic Acid in Fresh-Cut Potato. Food Chem. 2023, 410, 135444. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafez, O.H.; Othman, E.M.; Fahim, J.R.; Desoukey, S.Y.; Pimentel-Elardo, S.M.; Nodwell, J.R.; Schirmeister, T.; Tawfike, A.; Abdelmohsen, U.R. Metabolomics Analysis and Biological Investigation of Three Malvaceae Plants. Phytochem. Anal. 2020, 31, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Zhou, B.; Peng, Z.; Yao, M.; Wu, J.; Wu, X.; Guan, C.; Guan, M. Tissue-Specific Transcriptome and Metabolome Analysis Reveals the Response Mechanism of Brassica napus to Waterlogging Stress. Int. J. Mol. Sci. 2023, 24, 6015. [Google Scholar] [CrossRef]
- Cid, G.A.; Francioli, D.; Kolb, S.; Tandron Moya, Y.A.; von Wirén, N.; Hajirezaei, M.-R. Elucidating the Systemic Response of Wheat Plants under Waterlogging Based on Transcriptomic and Metabolic Approaches. J. Exp. Bot. 2023, 75, erad453. [Google Scholar] [CrossRef]
- Shang, P.; Shen, B.; Zeng, B.; Bi, L.; Qu, M.; Zheng, Y.; Ye, Y.; Li, W.; Zhou, X.; Yang, X.; et al. Integrated Transcriptomic and Metabolomics Analysis of the Root Responses of Orchardgrass to Submergence Stress. Int. J. Mol. Sci. 2023, 24, 2089. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Shah, S.; Kumar, R.; Vashisth, D.; Akhtar, M.Q.; Kumar, A.; Dwivedi, U.N.; Shasany, A.K. Ocimum Metabolomics in Response to Abiotic Stresses: Cold, Flood, Drought and Salinity. PLoS ONE 2019, 14, e0210903. [Google Scholar] [CrossRef]
- Gedam, P.A.; Khandagale, K.; Shirsat, D.; Thangasamy, A.; Kulkarni, O.; Kulkarni, A.; Patil, S.S.; Barvkar, V.T.; Mahajan, V.; Gupta, A.J.; et al. Elucidating the Molecular Responses to Waterlogging Stress in Onion (Allium cepa L.) Leaf by Comparative Transcriptome Profiling. Front. Plant Sci. 2023, 14, 1150909. [Google Scholar] [CrossRef]
- Sweetlove, L.J.; Beard, K.F.M.; Nunes-Nesi, A.; Fernie, A.R.; Ratcliffe, R.G. Not Just a Circle: Flux Modes in the Plant TCA Cycle. Trends Plant Sci. 2010, 15, 462–470. [Google Scholar] [CrossRef]
- Kumar, M.; Kumar Patel, M.; Kumar, N.; Bajpai, A.B.; Siddique, K.H.M. Metabolomics and Molecular Approaches Reveal Drought Stress Tolerance in Plants. Int. J. Mol. Sci. 2021, 22, 9108. [Google Scholar] [CrossRef]
- Czernicka, M.; Kęska, K.; Planchon, S.; Kapusta, M.; Popielarska-Konieczna, M.; Wesołowski, W.; Szklarczyk, M.; Renaut, J. Proteomic Studies of Roots in Hypoxia-Sensitive and -Tolerant Tomato Accessions Reveal Candidate Proteins Associated with Stress Priming. Cells 2022, 11, 500. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhan, J.; Luo, Z.; Zeng, N.; Zhang, W.; Zhang, H.; Li, L. Quantitative Proteomics and Relative Enzymatic Activities Reveal Different Mechanisms in Two Peanut Cultivars (Arachis hypogaea L.) Under Waterlogging Conditions. Front. Plant Sci. 2021, 12, 716114. [Google Scholar] [CrossRef]
- Alam, I.; Sharmin, S.A.; Kim, K.-H.; Kim, Y.-G.; Lee, J.J.; Bahk, J.D.; Lee, B.-H. Comparative Proteomic Approach to Identify Proteins Involved in Flooding Combined with Salinity Stress in Soybean. Plant Soil 2011, 346, 45–62. [Google Scholar] [CrossRef]
- Zhou, X.; Gong, F.; Dong, J.; Lin, X.; Cao, K.; Xu, H.; Zhou, X. Abscisic Acid Affects Phenolic Acid Content to Increase Tolerance to UV-B Stress in Rhododendron chrysanthum Pall. Int. J. Mol. Sci. 2024, 25, 1234. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Wu, C.; Liu, B.-Y.; Liang, H.-L.; Wang, M.-L.; Li, H. Transcriptomic and Metabolomic Profiling Reveals the Drought Tolerance Mechanism of Illicium difengpi (Schisandraceae). Front. Plant Sci. 2023, 14, 1284135. [Google Scholar] [CrossRef]
- Ojeda-Rivera, J.O.; Ulloa, M.; Pérez-Zavala, F.G.; Nájera-González, H.-R.; Roberts, P.A.; Yong-Villalobos, L.; Yadav, H.; Chávez Montes, R.A.; Herrera-Estrella, L.; Lopez-Arredondo, D. Enhanced Phenylpropanoid Metabolism Underlies Resistance to Fusarium oxysporum f. Sp. vasinfectum Race 4 Infection in the Cotton Cultivar Pima-S6 (Gossypium barbadense L.). Front. Genet. 2023, 14, 1271200. [Google Scholar] [CrossRef]
- Zhang, S.; Qi, X.; Zhu, R.; Ye, D.; Shou, M.; Peng, L.; Qiu, M.; Shi, M.; Kai, G. Transcriptome Analysis of Salvia miltiorrhiza under Drought Stress. Plants 2024, 13, 161. [Google Scholar] [CrossRef]
- La, V.H.; Tran, D.H.; Han, V.-C.; Nguyen, T.D.; Duong, V.C.; Nguyen, V.H.; Tran, A.T.; Nguyen, T.H.G.; Ngo, X.B. Drought Stress-Responsive Abscisic Acid and Salicylic Acid Crosstalk with the Phenylpropanoid Pathway in Soybean Seeds. Physiol. Plant 2023, 175, e14050. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Li, X.; Yang, Q.; Zhao, M.; Bai, W.; Liang, Y.; Liu, X.; Gao, B.; Zhang, D. Transcriptional Profiling Analysis Providing Insights into Desiccation Tolerance Mechanisms of the Desert Moss Syntrichia caninervis. Front. Plant Sci. 2023, 14, 1127541. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Su, Y.; Wang, T. Full-Length Transcriptome Analysis of Four Different Tissues of Cephalotaxus oliveri. Int. J. Mol. Sci. 2021, 22, 787. [Google Scholar] [CrossRef]
- Balfagón, D.; Pascual, L.S.; Sengupta, S.; Halliday, K.J.; Gómez-Cadenas, A.; Peláez-Vico, M.Á.; Sinha, R.; Mittler, R.; Zandalinas, S.I. WRKY48 Negatively Regulates Plant Acclimation to a Combination of High Light and Heat Stress. Plant J. 2024, 117, 1642–1655. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hu, L.; Yu, L.; Liang, S.; Qu, H.; Wang, M.; Hao, Z.; Yang, L.; Shi, J.; Chen, J. Physiological and Transcriptomic Analysis Revealed That the Accumulation of Reactive Oxygen Species Caused the Low Temperature Sensitivity of Liriodendron × Sinoamericanum. Plant Sci. 2024, 342, 112020. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, J.; Qu, G.; Chen, S. The Cold-Responsive C-Repeat Binding Factors in Betula Platyphylla Suk. Positively Regulate Cold Tolerance. Plant Sci. 2024, 341, 112012. [Google Scholar] [CrossRef]
- Yoon, Y.; Seo, D.H.; Shin, H.; Kim, H.J.; Kim, C.M.; Jang, G. The Role of Stress-Responsive Transcription Factors in Modulating Abiotic Stress Tolerance in Plants. Agronomy 2020, 10, 788. [Google Scholar] [CrossRef]
- Zhang, H.; Li, G.; Yan, C.; Zhang, X.; Cao, N.; Le, M.; Hu, X.; Zhu, F.; Liu, W. Elucidating the Molecular Responses to Waterlogging Stress in Cucumis melo by Comparative Transcriptome Profiling. Horticulturae 2022, 8, 891. [Google Scholar] [CrossRef]
- Wei, H.; Chen, J.; Zhang, X.; Lu, Z.; Lian, B.; Liu, G.; Chen, Y.; Zhong, F.; Yu, C.; Zhang, J. Comprehensive Analysis of Annexin Gene Family and Its Expression in Response to Branching Architecture and Salt Stress in Crape Myrtle. BMC Plant Biol. 2024, 24, 78. [Google Scholar] [CrossRef]
- Tian, H.; Fan, G.; Xiong, X.; Wang, H.; Zhang, S.; Geng, G. Characterization and Transformation of the CabHLH18 Gene from Hot Pepper to Enhance Waterlogging Tolerance. Front. Plant Sci. 2023, 14, 1285198. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Zhou, M.; Hu, Y.; Yuan, J. PacBio Full-Length Sequencing Integrated with RNA-Seq Reveals the Molecular Mechanism of Waterlogging and Its Recovery in Paeonia ostii. Front. Plant Sci. 2022, 13, 1030584. [Google Scholar] [CrossRef] [PubMed]
- Borrego-Benjumea, A.; Carter, A.; Tucker, J.R.; Yao, Z.; Xu, W.; Badea, A. Genome-Wide Analysis of Gene Expression Provides New Insights into Waterlogging Responses in Barley (Hordeum vulgare L.). Plants 2020, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Regon, P.; Dey, S.; Rehman, M.; Pradhan, A.K.; Chowra, U.; Tanti, B.; Talukdar, A.D.; Panda, S.K. Transcriptomic Analysis Revealed Reactive Oxygen Species Scavenging Mechanisms Associated with Ferrous Iron Toxicity in Aromatic Keteki Joha Rice. Front. Plant Sci. 2022, 13, 798580. [Google Scholar] [CrossRef]
- Yin, J.; Wang, L.; Zhao, J.; Li, Y.; Huang, R.; Jiang, X.; Zhou, X.; Zhu, X.; He, Y.; He, Y.; et al. Genome-Wide Characterization of the C2H2 Zinc-Finger Genes in Cucumis sativus and Functional Analyses of Four CsZFPs in Response to Stresses. BMC Plant Biol. 2020, 20, 359. [Google Scholar] [CrossRef]
- Li, Z.; Bai, D.; Zhong, Y.; Lin, M.; Sun, L.; Qi, X.; Hu, C.; Fang, J. Full-Length Transcriptome and RNA-Seq Analyses Reveal the Mechanisms Underlying Waterlogging Tolerance in Kiwifruit (Actinidia valvata). Int. J. Mol. Sci. 2022, 23, 3237. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, S.; Liu, H.; Hu, Q.; Liu, X.; Wang, J.; Wang, J.; Xin, W.; Chen, Q. Physiological and Transcriptomic Analysis of the Mangrove Species Kandelia obovata in Response to Flooding Stress. Mar. Pollut. Bull. 2023, 196, 115598. [Google Scholar] [CrossRef]
- Han, G.; Qiao, Z.; Li, Y.; Wang, C.; Wang, B. The Roles of CCCH Zinc-Finger Proteins in Plant Abiotic Stress Tolerance. Int. J. Mol. Sci. 2021, 22, 8327. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Liao, K.; Wang, L.-N.; Shi, L.-L.; Zhang, Y.; Xu, L.-J.; Zhou, Y.; Li, J.-F.; Chen, Y.-Q.; Chen, Q.-F.; et al. Calcium-Dependent Activation of CPK12 Facilitates Its Cytoplasm-to-Nucleus Translocation to Potentiate Plant Hypoxia Sensing by Phosphorylating ERF-VII Transcription Factors. Mol. Plant 2023, 16, 979–998. [Google Scholar] [CrossRef]
- Zhou, Y.; Tan, W.-J.; Xie, L.-J.; Qi, H.; Yang, Y.-C.; Huang, L.-P.; Lai, Y.-X.; Tan, Y.-F.; Zhou, D.-M.; Yu, L.-J.; et al. Polyunsaturated Linolenoyl-CoA Modulates ERF-VII-Mediated Hypoxia Signaling in Arabidopsis. J. Integr. Plant Biol. 2020, 62, 330–348. [Google Scholar] [CrossRef] [PubMed]

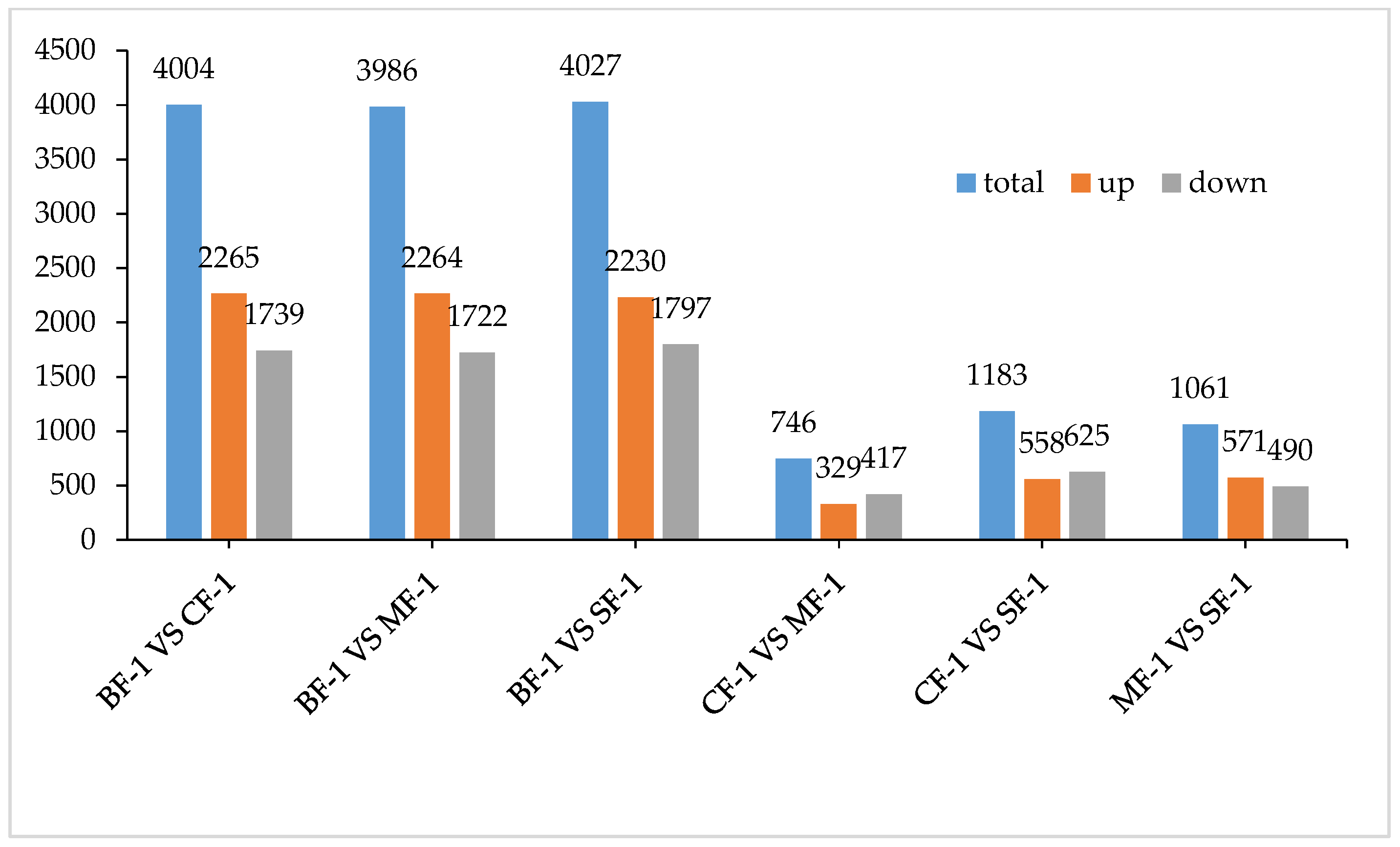



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
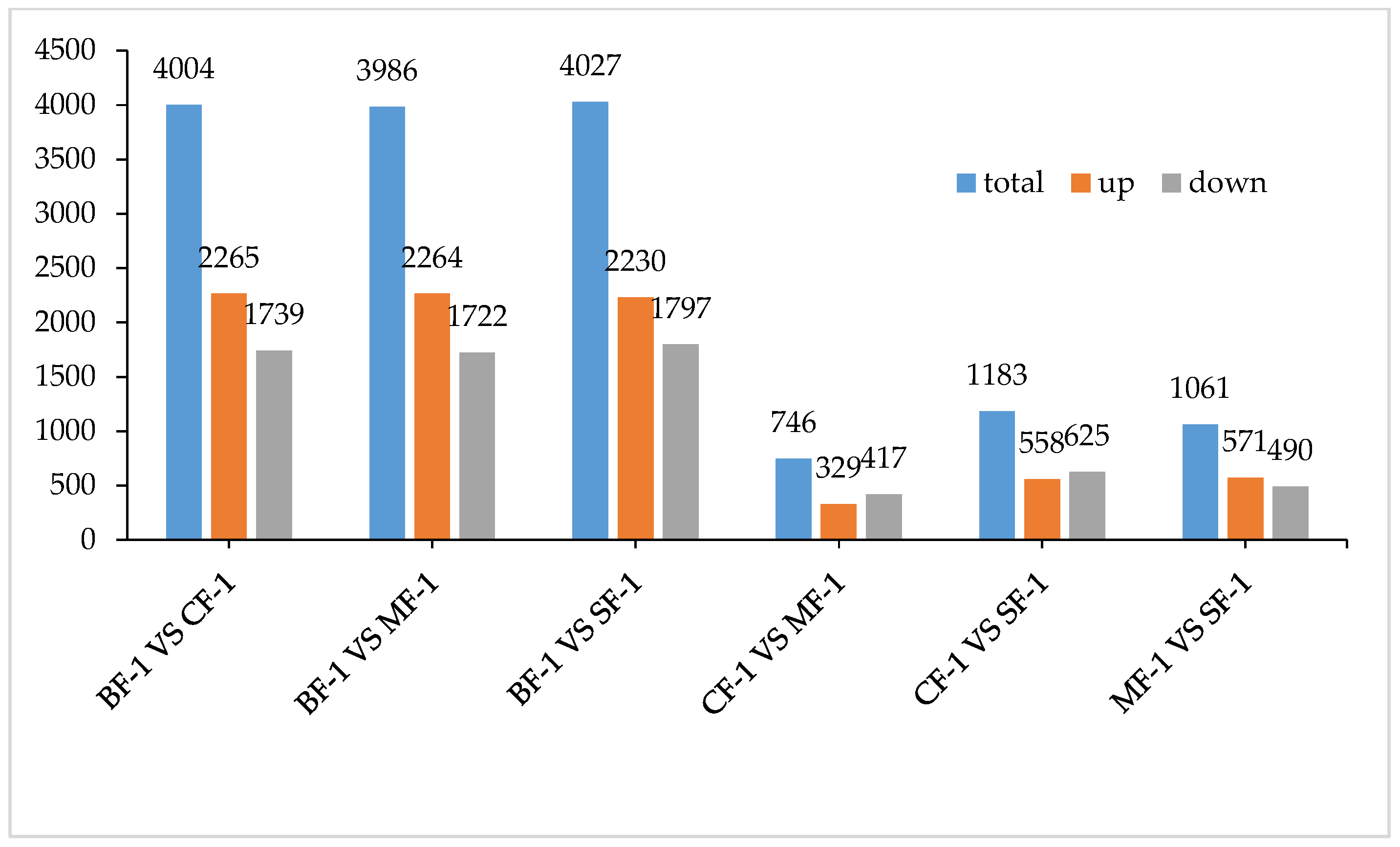{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5’-3’) |
|---|---|
| actin-F | ATTCCCAAGGCAGCCACAA |
| actin-R | ATACAGACACCCAGCCTCCTTTA |
| DN2723101-F | CGTTCCTCAACAACTCCCAA |
| DN2723101-R | GAAGAAAAGAAGTCAGTGTAATCCC |
| DN914502-F | TTCACATGTATGCTGCTAACAAGTC |
| DN914502-R | CAGGTAAGTCTACTGCAAAACGAAA |
| DN3612101-F | CATAGGCACAACCACGGCA |
| DN3612101-R | AATTTTCACCGTCCCAGCC |
| DN496621-F | TAGGCCGACAATATTTCTAGAGATC |
| DN496621-R | AATCCTCCACATCCGCCTT |
| DN122401-F | ACCACCATCTCATCAAACTCCTCA |
| DN122401-R | ATACAGCGTGAAGGTCAAGTCCC |
| DN1086201-F | CAAACCTAGAGTGCATGACCATAAT |
| DN1086201-R | GAGGGATTTTATCTCCATAGCGA |
| Cluster | Compounds | Molecular Formula | Molecular Weight (g/mol) | HMDB_taxonomy |
|---|---|---|---|---|
| cluster3 | Homoharringtonine | C29H39NO9 | 545.62 | Cephalotaxus alkaloids |
| cluster3 | Apiin | C26H28O14 | 564.49 | Naphthalenes |
| cluster3 | All-trans-phytofluene | C40H62 | 542.92 | Prenol lipids |
| cluster3 | Pelargonidin 3-O-beta-D-sambubioside | C26H29O14 | 565.50 | -- |
| cluster5 | Copal-8-ol diphosphate | C20H38O8P2 | 468.50 | -- |
| cluster5 | Ganoderenic acid A | C30H42O7 | 514.65 | Prenol lipids |
| cluster5 | Solamargine | C45H73NO15 | 868.06 | Azaspirodecane derivatives |
| cluster5 | OA-6129 A | C20H31N3O7S | 457.55 | -- |
| cluster5 | O-Phospho-L-homoserine | C4H10NO6P | 199.02 | Carboxylic acids and derivatives |
| cluster5 | (3S,2′S)-4-Ketomyxol 2′-alpha-L-fucoside | C46H64O8 | 745.00 | -- |
| cluster5 | Bacoside A3 | C47H76O18 | 929.10 | -- |
| cluster5 | Schaftoside 4′-O-glucoside | C32H38O19 | 726.60 | Flavonoids |
| cluster5 | Pelargonin | C27H31O15 | 595.526 | Flavonoids |
| cluster5 | Muzanzagenin | C27H38O5 | 442.60 | Steroids and steroid derivatives |
| cluster5 | Amaranthin | C30H34N2O19 | 726.59 | Betalains |
| cluster5 | DL-4-Hydroxy-2-ketoglutarate | C5H6O6 | 162.10 | -- |
| cluster5 | PG(16:0/0:0) | C22H45O9P | 484.60 | -- |
| cluster5 | D-Maltose | C12H22O11 | 342.30 | Organooxygen compounds |
| cluster5 | 3.alpha.-Mannobiose | C12H22O11 | 342.30 | -- |
| cluster5 | Chelidonic acid | C7H4O6 | 184.10 | Pyrans |
| cluster5 | ω-Pentadecalactone | C15H28O2 | 240.38 | -- |
| cluster5 | Gypenoside XVII | C48H82O18 | 947.20 | Prenol lipids |
| cluster5 | PG(20:2(11Z,14Z)/16:1(9Z)) | C42H77O10P | 773.00 | -- |
| cluster5 | Dioctyl phthalate | C24H38O4 | 390.60 | Benzene and substituted derivatives |
| cluster5 | DG(18:3(9Z,12Z,15Z)/18:3(9Z,12Z,15Z)/0:0) | C39H64O5 | 612.90 | Fatty Acyls |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, T.; Zhou, X.; Lin, J.; Zhou, X.; Wang, H.; Chen, T. Transcriptomic and Metabolomic Analyses Reveal the Response Mechanism of Ophiopogon japonicus to Waterlogging Stress. Biology 2024, 13, 197. https://doi.org/10.3390/biology13030197
Cheng T, Zhou X, Lin J, Zhou X, Wang H, Chen T. Transcriptomic and Metabolomic Analyses Reveal the Response Mechanism of Ophiopogon japonicus to Waterlogging Stress. Biology. 2024; 13(3):197. https://doi.org/10.3390/biology13030197
Chicago/Turabian StyleCheng, Tingting, Xia Zhou, Juan Lin, Xianjian Zhou, Hongsu Wang, and Tiezhu Chen. 2024. "Transcriptomic and Metabolomic Analyses Reveal the Response Mechanism of Ophiopogon japonicus to Waterlogging Stress" Biology 13, no. 3: 197. https://doi.org/10.3390/biology13030197