
Engineering a Dual Specificity γδ T-Cell Receptor for Cancer Immunotherapy

 , ,
, , 
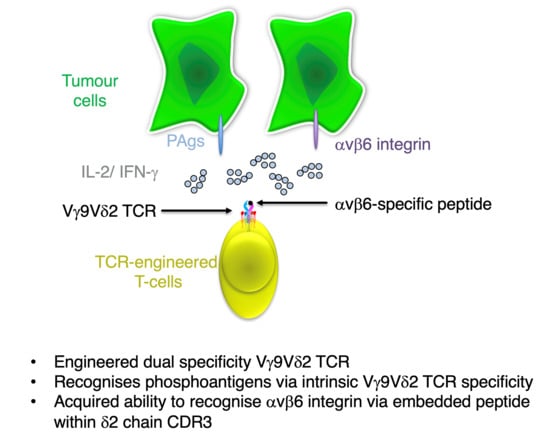
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Human Samples
2.3. Retroviral Constructs
2.4. Production of Retroviral Vector
2.5. T-Cell Activation
2.6. T-Cell Transduction
2.7. FACS Analysis
2.8. Stimulation of Engineered T-Cells on Immobilised αvβ6 Integrin
2.9. Enzyme-Linked Immunosorbent Assay
2.10. Cytotoxicity Assays
2.11. Statistical Analysis
3. Results
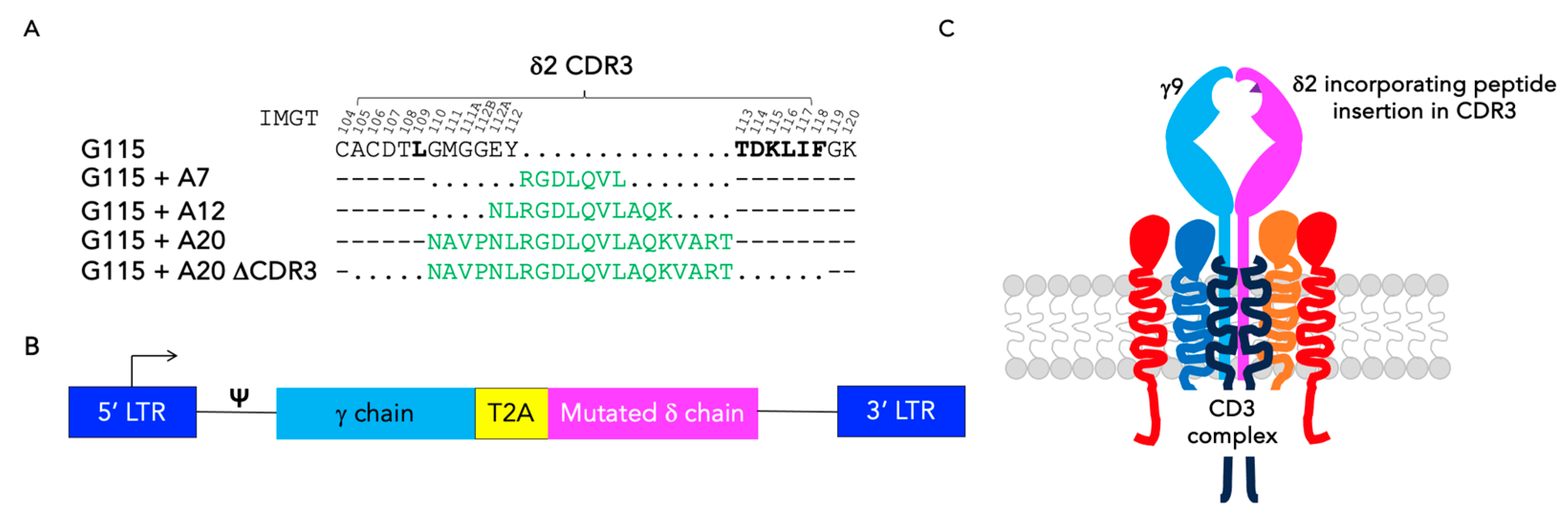
3.1. Engineering of CDR3 Mutant Derivatives of the G115 γδ TCR
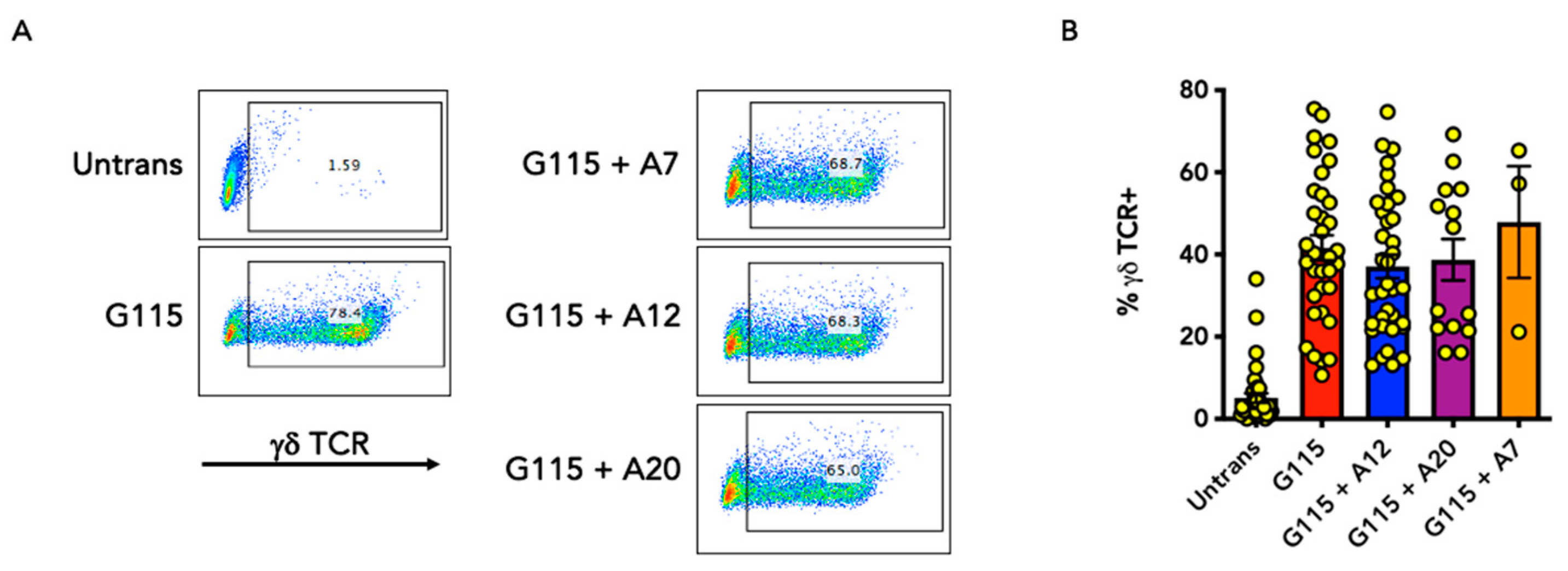
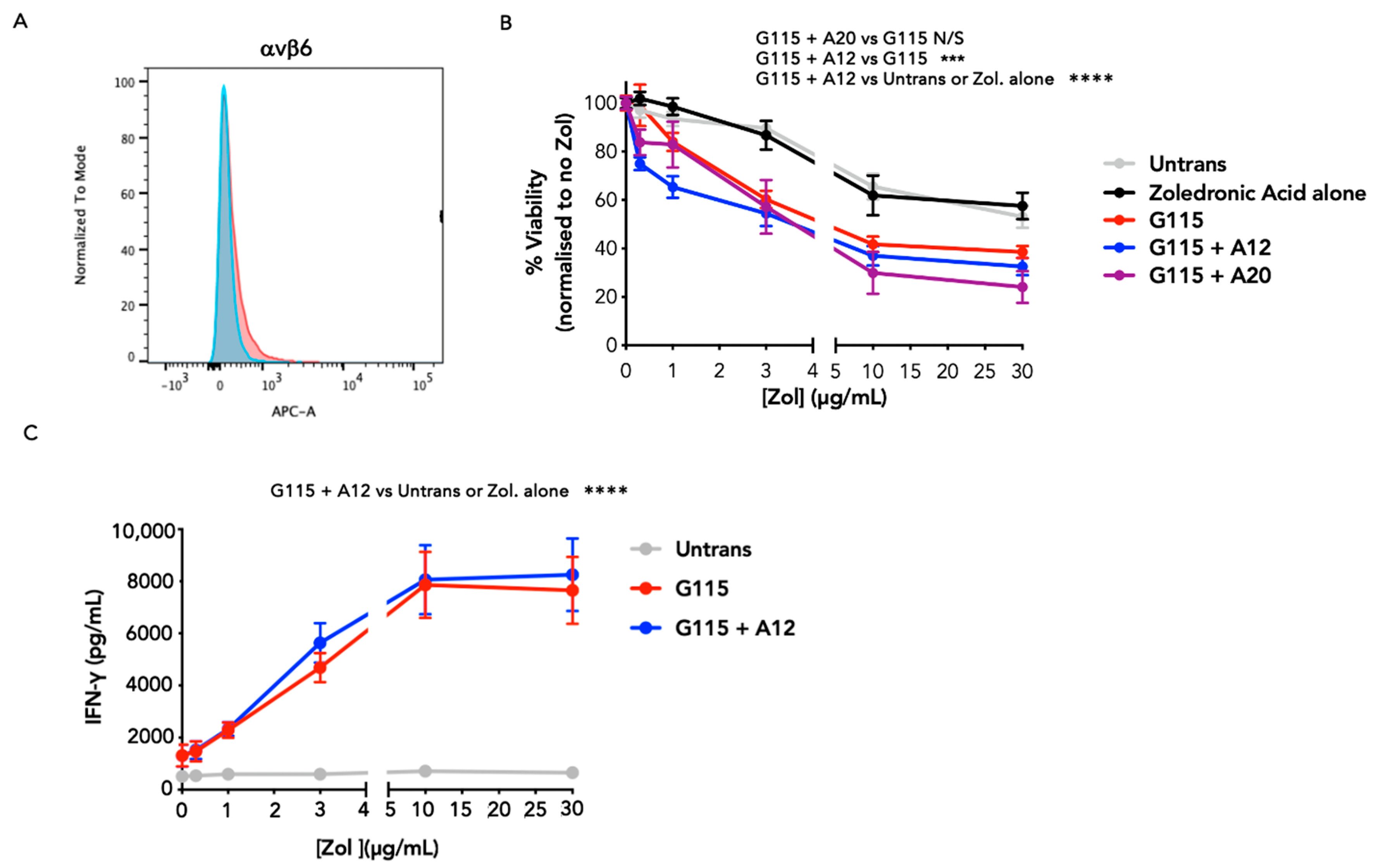
3.2. Evaluation of αvβ6 Integrin Specificity of Engineered G115 γδ TCRs
3.3. Evaluation of PAg Specificity of Engineered G115 γδ TCRs on K562 Cells
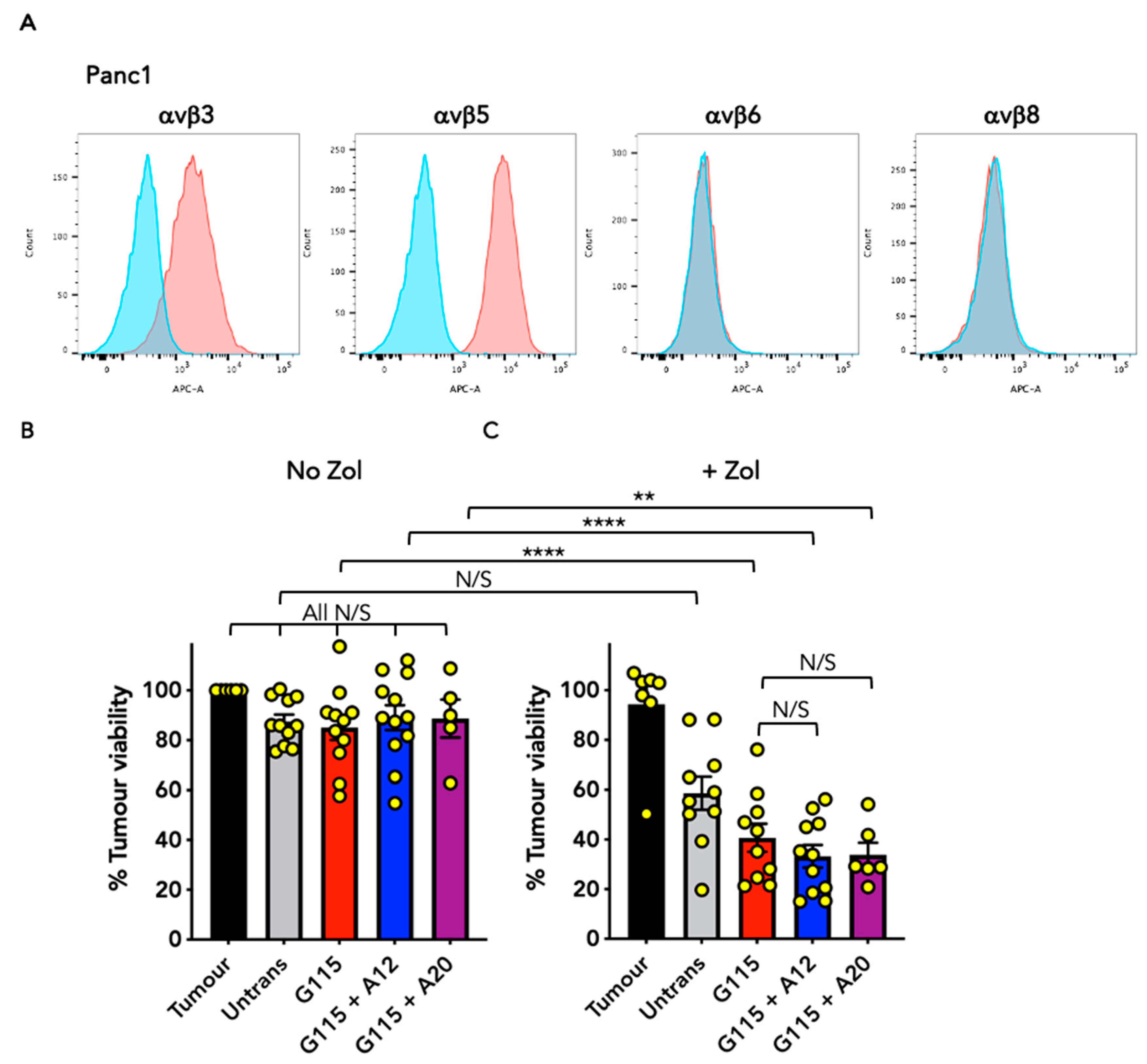
3.4. Evaluation of Anti-Tumour Activity of Engineered G115 γδ TCRs on Pancreatic Tumour Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with gammadelta T cells: Many paths ahead of us. Cell Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, Z.; Prinz, I.; Dechanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (gammadelta) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Herrmann, T.; Fichtner, A.S.; Karunakaran, M.M. An Update on the Molecular Basis of Phosphoantigen Recognition by Vgamma9Vdelta2 T Cells. Cells 2020, 9, 1433. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Ma, X.; Yang, Y.; Qu, Y.; Li, X.; Zhu, X.; Ma, W.; Duan, J.; Xue, J.; Yang, H.; et al. Phosphoantigens glue butyrophilin 3A1 and 2A1 to activate Vgamma9Vdelta2 T cells. Nature 2023, 621, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Recio, C.; Aranda-Tavio, H.; Guerra-Rodriguez, M.; Garcia-Castellano, J.M.; Fernandez-Perez, L. The Mevalonate Pathway, a Metabolic Target in Cancer Therapy. Front. Oncol. 2021, 11, 626971. [Google Scholar] [CrossRef]
- Saura-Esteller, J.; de Jong, M.; King, L.A.; Ensing, E.; Winograd, B.; de Gruijl, T.D.; Parren, P.; van der Vliet, H.J. Gamma Delta T-Cell Based Cancer Immunotherapy: Past-Present-Future. Front. Immunol. 2022, 13, 915837. [Google Scholar] [CrossRef]
- Marcu-Malina, V.; Heijhuurs, S.; van Buuren, M.; Hartkamp, L.; Strand, S.; Sebestyen, Z.; Scholten, K.; Martens, A.; Kuball, J. Redirecting alphabeta T cells against cancer cells by transfer of a broadly tumor-reactive gammadeltaT-cell receptor. Blood 2011, 118, 50–59. [Google Scholar] [CrossRef]
- Vyborova, A.; Beringer, D.X.; Fasci, D.; Karaiskaki, F.; van Diest, E.; Kramer, L.; de Haas, A.; Sanders, J.; Janssen, A.; Straetemans, T.; et al. gamma9delta2T cell diversity and the receptor interface with tumor cells. J. Clin. Investig. 2020, 130, 4637–4651. [Google Scholar] [CrossRef]
- Hernandez-Lopez, P.; van Diest, E.; Brazda, P.; Heijhuurs, S.; Meringa, A.; Hoorens van Heyningen, L.; Riillo, C.; Schwenzel, C.; Zintchenko, M.; Johanna, I.; et al. Dual targeting of cancer metabolome and stress antigens affects transcriptomic heterogeneity and efficacy of engineered T cells. Nat. Immunol. 2024, 25, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Grunder, C.; van Dorp, S.; Hol, S.; Drent, E.; Straetemans, T.; Heijhuurs, S.; Scholten, K.; Scheper, W.; Sebestyen, Z.; Martens, A.; et al. gamma9 and delta2CDR3 domains regulate functional avidity of T cells harboring gamma9delta2TCRs. Blood 2012, 120, 5153–5162. [Google Scholar] [CrossRef] [PubMed]
- DiCara, D.; Rapisarda, C.; Sutcliffe, J.L.; Violette, S.M.; Weinreb, P.H.; Hart, I.R.; Howard, M.J.; Marshall, J.F. Structure-function analysis of Arg-Gly-Asp helix motifs in alpha v beta 6 integrin ligands. J. Biol. Chem. 2007, 282, 9657–9665. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.M.; Vallath, S.; Maher, J. The integrin alphavbeta6: A novel target for CAR T-cell immunotherapy? Biochem. Soc. Trans. 2016, 44, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.M.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Petrovic, R.M.G.; Kao, Y.V.; Saxena, S.A.; Romain, A.; Costa-Guerra, J.A.; Violette, S.; et al. Targeting of Aberrant alphavbeta6 Integrin Expression in Solid Tumors Using Chimeric Antigen Receptor-Engineered T Cells. Mol. Ther. 2017, 25, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Whilding, L.M.; Halim, L.; Draper, B.; Parente-Pereira, A.C.; Zabinski, T.; Davies, D.M.; Maher, J. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers 2019, 11, 674. [Google Scholar] [CrossRef]
- Phanthaphol, N.; Somboonpatarakun, C.; Suwanchiwasiri, K.; Chieochansin, T.; Sujjitjoon, J.; Wongkham, S.; Maher, J.; Junking, M.; Yenchitsomanus, P.T. Chimeric Antigen Receptor T Cells Targeting Integrin alphavbeta6 Expressed on Cholangiocarcinoma Cells. Front. Oncol. 2021, 11, 657868. [Google Scholar] [CrossRef] [PubMed]
- Kogelberg, H.; Tolner, B.; Thomas, G.J.; Di Cara, D.; Minogue, S.; Ramesh, B.; Sodha, S.; Marsh, D.; Lowdell, M.W.; Meyer, T.; et al. Engineering a single-chain Fv antibody to alpha v beta 6 integrin using the specificity-determining loop of a foot-and-mouth disease virus. J. Mol. Biol. 2008, 382, 385–401. [Google Scholar] [CrossRef]
- Lefranc, M.P. IMGT, the international ImMunoGeneTics database. Nucleic Acids Res. 2003, 31, 307–310. [Google Scholar] [CrossRef]
- Karunakaran, M.M.; Gobel, T.W.; Starick, L.; Walter, L.; Herrmann, T. Vgamma9 and Vdelta2 T cell antigen receptor genes and butyrophilin 3 (BTN3) emerged with placental mammals and are concomitantly preserved in selected species like alpaca (Vicugna pacos). Immunogenetics 2014, 66, 243–254. [Google Scholar] [CrossRef]
- Larcombe-Young, D.; Whilding, L.; Davies, D.M.; Draper, B.; Bechman, N.; Maher, J. Generation of human parallel chimeric antigen receptor (pCAR) T cells to achieve synergistic T cell co-stimulation. STAR Protoc. 2022, 3, 101414. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Xu, W.; Goolia, M.; Zhang, Z. Characterization of monoclonal antibodies against foot-and-mouth disease virus serotype O and application in identification of antigenic variation in relation to vaccine strain selection. Virol. J. 2014, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Kraft, S.; Diefenbach, B.; Mehta, R.; Jonczyk, A.; Luckenbach, G.A.; Goodman, S.L. Definition of an unexpected ligand recognition motif for alphav beta6 integrin. J. Biol. Chem. 1999, 274, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- D’Asaro, M.; La Mendola, C.; Di Liberto, D.; Orlando, V.; Todaro, M.; Spina, M.; Guggino, G.; Meraviglia, S.; Caccamo, N.; Messina, A.; et al. V gamma 9V delta 2 T lymphocytes efficiently recognize and kill zoledronate-sensitized, imatinib-sensitive, and imatinib-resistant chronic myelogenous leukemia cells. J. Immunol. 2010, 184, 3260–3268. [Google Scholar] [CrossRef] [PubMed]
- Melandri, D.; Zlatareva, I.; Chaleil, R.A.G.; Dart, R.J.; Chancellor, A.; Nussbaumer, O.; Polyakova, O.; Roberts, N.A.; Wesch, D.; Kabelitz, D.; et al. The gammadeltaTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat. Immunol. 2018, 19, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Reijneveld, J.F.; Ocampo, T.A.; Shahine, A.; Gully, B.S.; Vantourout, P.; Hayday, A.C.; Rossjohn, J.; Moody, D.B.; Van Rhijn, I. Human gammadelta T cells recognize CD1b by two distinct mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 22944–22952. [Google Scholar] [CrossRef] [PubMed]
- Dolton, G.; Rius, C.; Wall, A.; Szomolay, B.; Bianchi, V.; Galloway, S.A.E.; Hasan, M.S.; Morin, T.; Caillaud, M.E.; Thomas, H.L.; et al. Targeting of multiple tumor-associated antigens by individual T cell receptors during successful cancer immunotherapy. Cell 2023, 186, 3333–3349 e3327. [Google Scholar] [CrossRef] [PubMed]
- Logan, D.; Abu-Ghazaleh, R.; Blakemore, W.; Curry, S.; Jackson, T.; King, A.; Lea, S.; Lewis, R.; Newman, J.; Parry, N.; et al. Structure of a major immunogenic site on foot-and-mouth disease virus. Nature 1993, 362, 566–568. [Google Scholar] [CrossRef]
- Morton, L.T.; Reijmers, R.M.; Wouters, A.K.; Kweekel, C.; Remst, D.F.G.; Pothast, C.R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Simultaneous Deletion of Endogenous TCRalphabeta for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol. Ther. 2020, 28, 64–74. [Google Scholar] [CrossRef]
- Heemskerk, M.H. T-cell receptor gene transfer for the treatment of leukemia and other tumors. Haematologica 2010, 95, 15–19. [Google Scholar] [CrossRef]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; Halladay, T.; Yang, L. Immune evasion in cell-based immunotherapy: Unraveling challenges and novel strategies. J. Biomed. Sci. 2024, 31, 5. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davies, D.M.; Pugliese, G.; Parente Pereira, A.C.; Whilding, L.M.; Larcombe-Young, D.; Maher, J. Engineering a Dual Specificity γδ T-Cell Receptor for Cancer Immunotherapy. Biology 2024, 13, 196. https://doi.org/10.3390/biology13030196
Davies DM, Pugliese G, Parente Pereira AC, Whilding LM, Larcombe-Young D, Maher J. Engineering a Dual Specificity γδ T-Cell Receptor for Cancer Immunotherapy. Biology. 2024; 13(3):196. https://doi.org/10.3390/biology13030196
Chicago/Turabian StyleDavies, David M., Giuseppe Pugliese, Ana C. Parente Pereira, Lynsey M. Whilding, Daniel Larcombe-Young, and John Maher. 2024. "Engineering a Dual Specificity γδ T-Cell Receptor for Cancer Immunotherapy" Biology 13, no. 3: 196. https://doi.org/10.3390/biology13030196
APA StyleDavies, D. M., Pugliese, G., Parente Pereira, A. C., Whilding, L. M., Larcombe-Young, D., & Maher, J. (2024). Engineering a Dual Specificity γδ T-Cell Receptor for Cancer Immunotherapy. Biology, 13(3), 196. https://doi.org/10.3390/biology13030196