

Formoterol Acting via β2-Adrenoreceptor Restores Mitochondrial Dysfunction Caused by Parkinson’s Disease-Related UQCRC1 Mutation and Improves Mitochondrial Homeostasis Including Dynamic and Transport
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. GFP-Labeled Mitochondria
2.3. Cell Number, Cell Viability, and Filopodia Outgrowth
2.4. Quantitative Mitochondrial Morphology and Density
2.5. Mitochondrial Function
2.6. Mitochondrial Motility
2.7. Western Blotting and Protein Phosphorylation Antibody Array
2.8. Protein Kinase A Activity
2.9. Statistics
3. Results
3.1. Formoterol Increased the Viability of UQCRC1 Mutant Cells and Protected against Tertiary-Butyl Hydroperoxide-Induced Cell Damage in a β2-Adrenoceptor-Dependent Manner
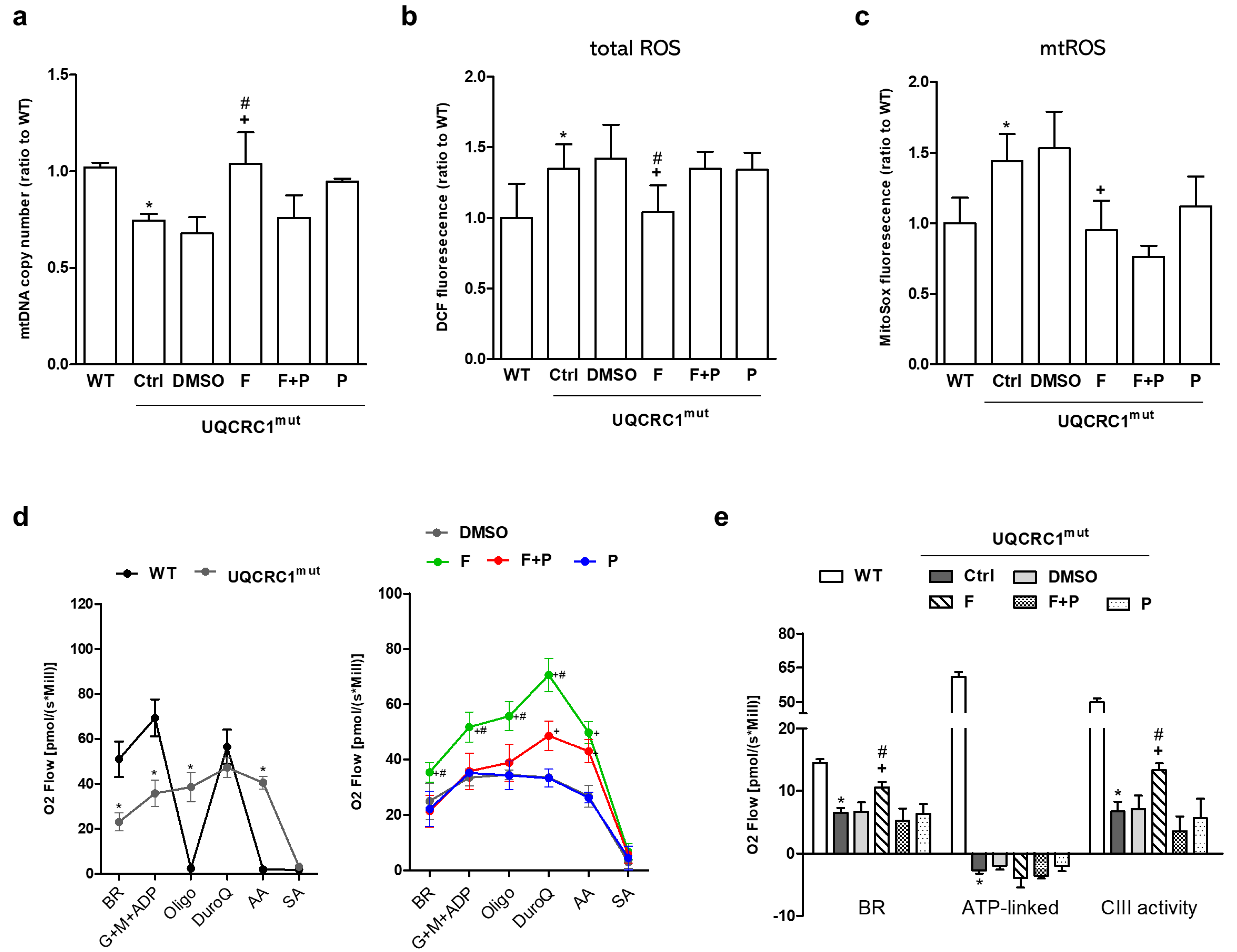
3.2. Formoterol Enhanced mtDNA Copy Number, Reduced ROS Levels, and Restored Mitochondrial Respiration and Complex III Activity, but Not ATP-Linked Respiration
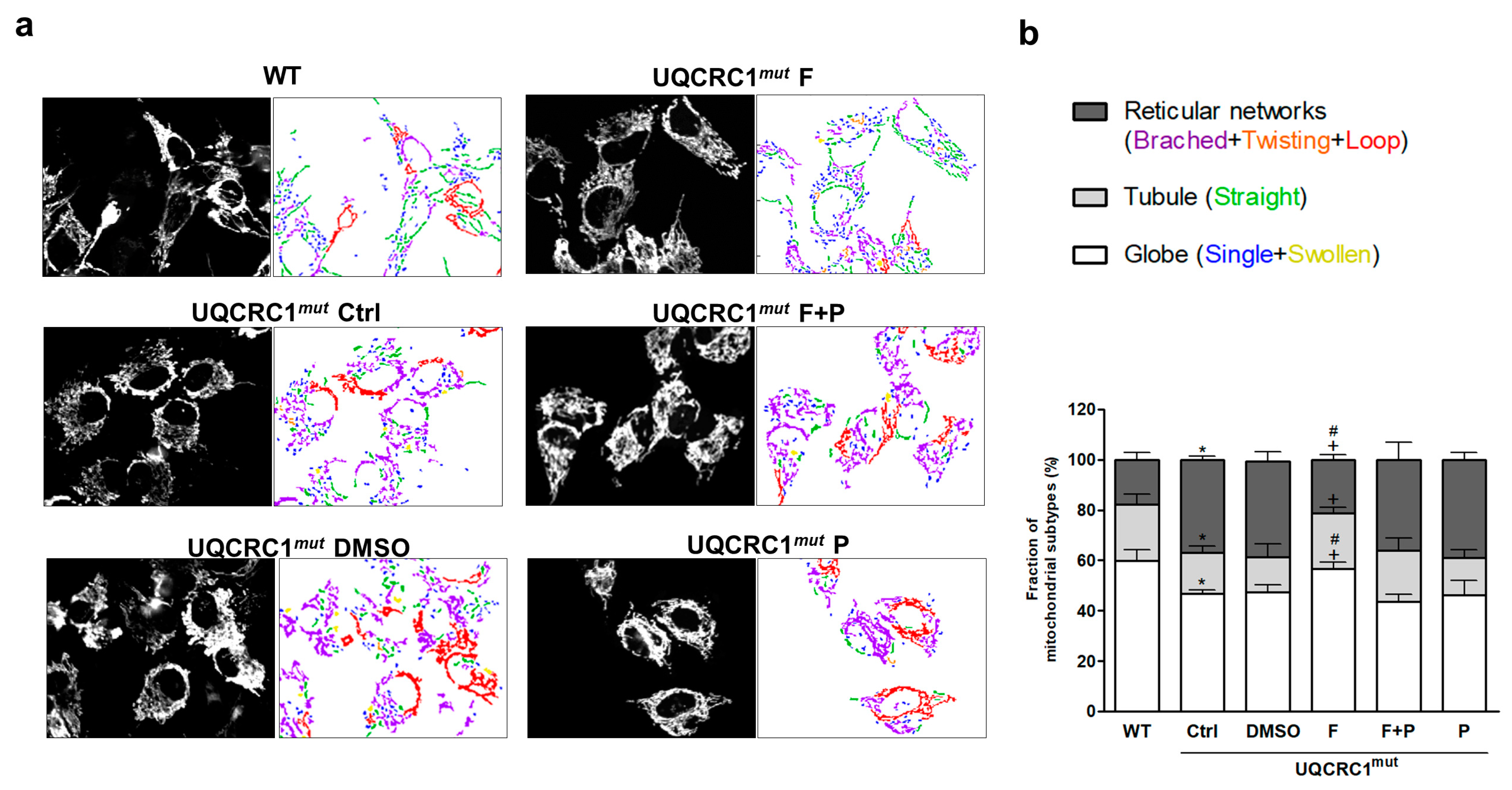
3.3. Formoterol Improved the Transition from an Abnormal, Clustered Network to a More Tubular and Globular Mitochondrial Morphology
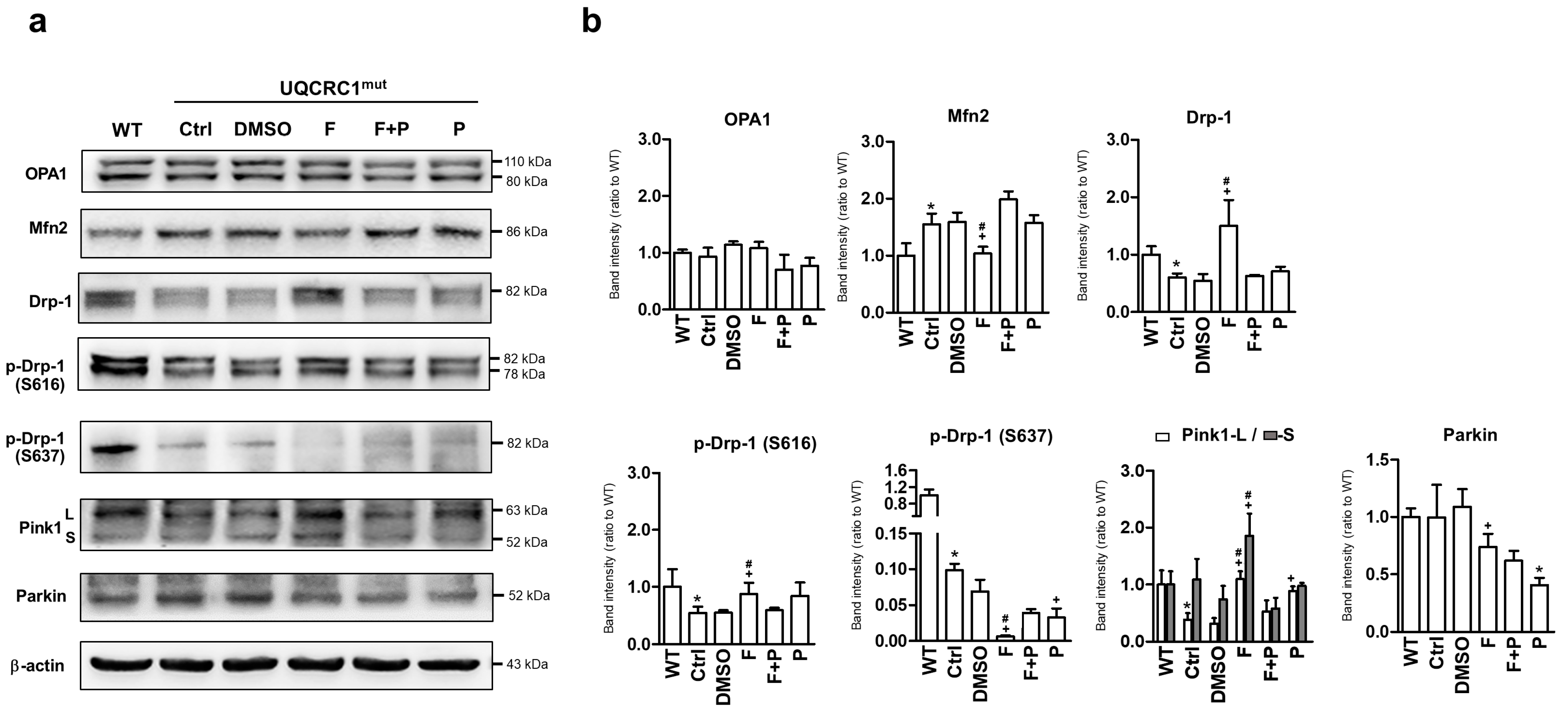
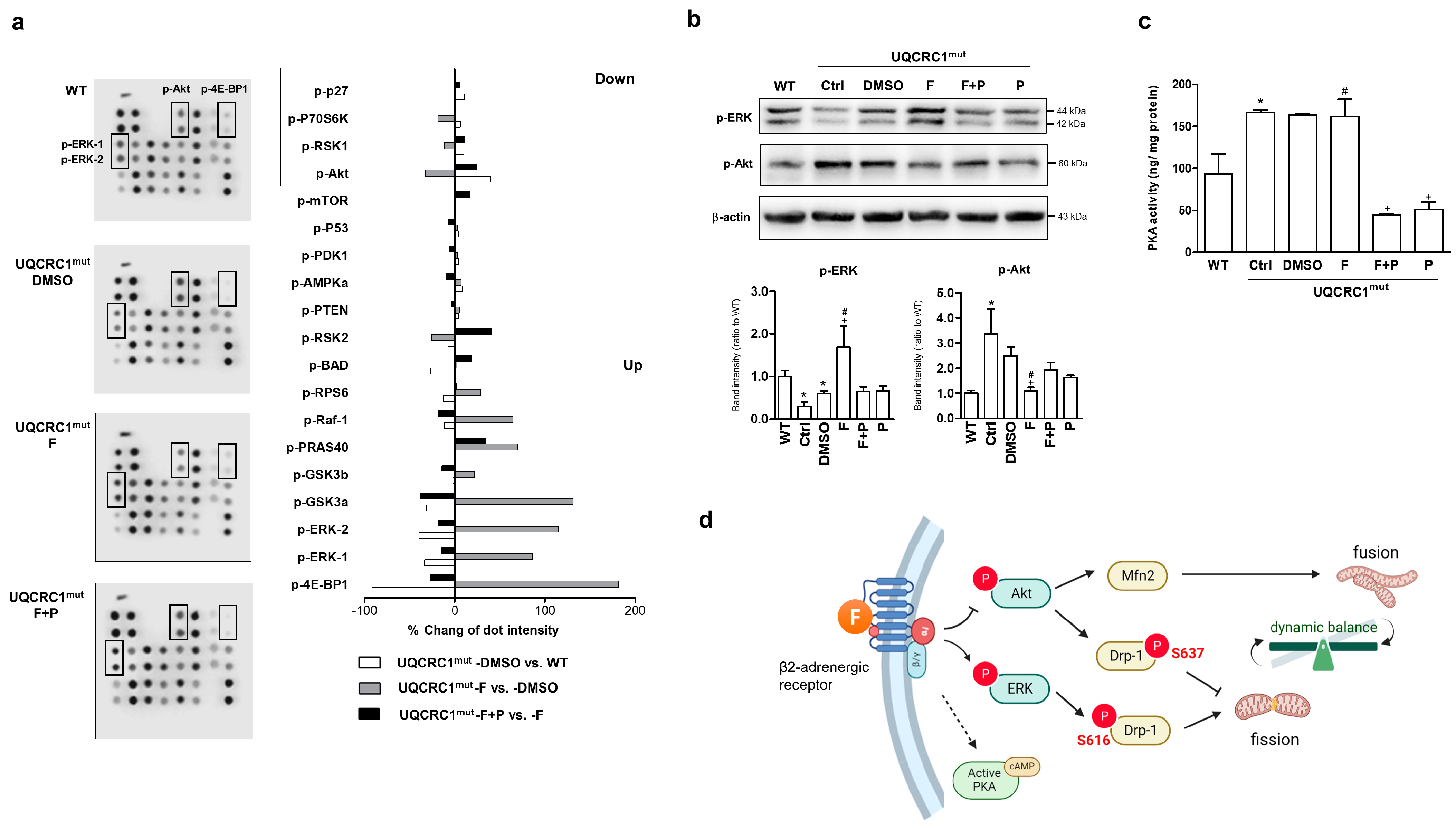
3.4. Formoterol Regulated Mitochondrial Fusion–Fission Balance through β2-Adrenoreceptor Activation and Was Linked to Counter-Regulation of ERK and Akt Signals
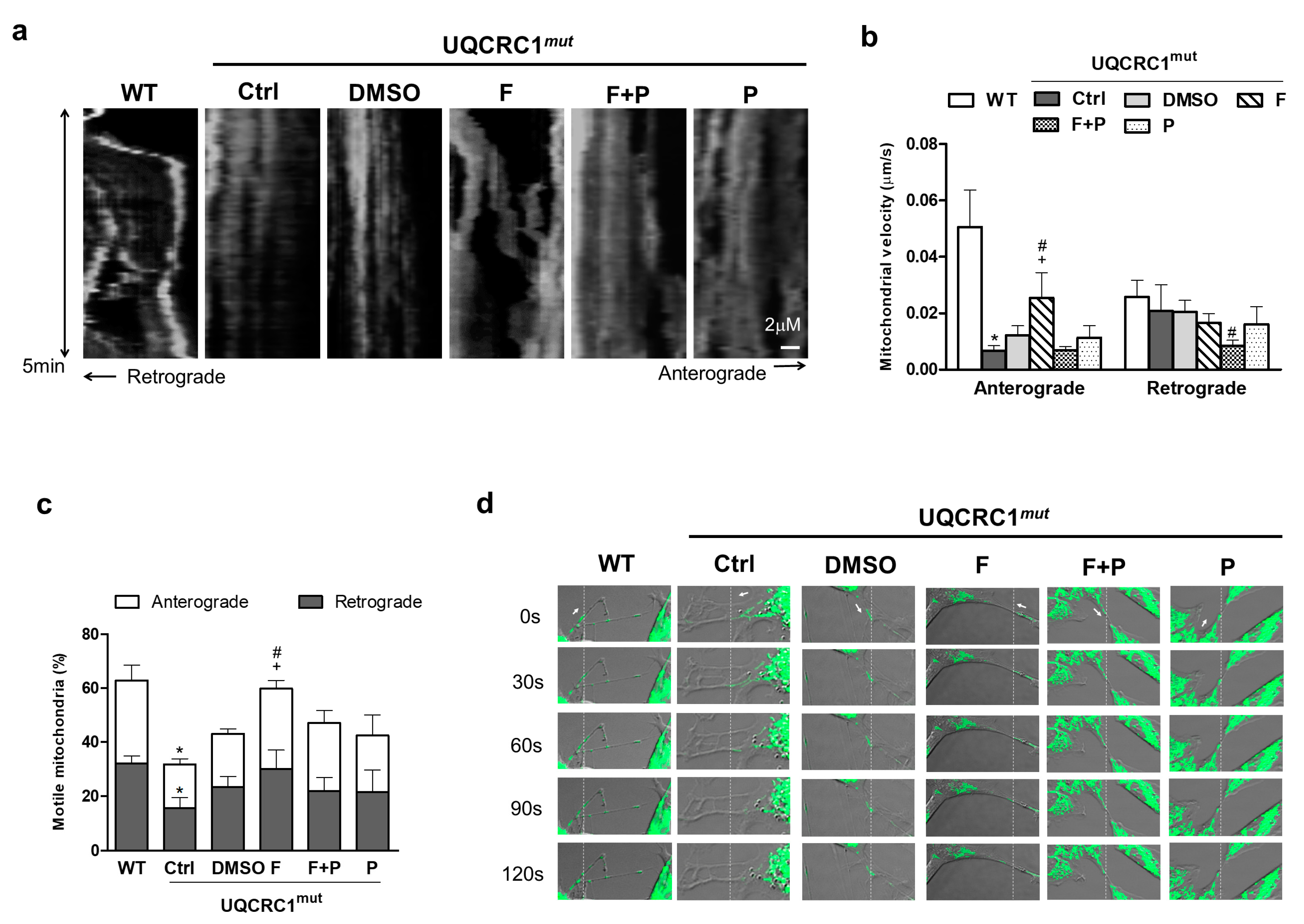
3.5. Formoterol Increased the Efficiency of Mitochondrial Anterograde Transportation and Its Mobility
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Billington, C.K.; Penn, R.B.; Hall, I.P. β2 Agonists. In Pharmacology and Therapeutics of Asthma and COPD; Springer: Cham, Switzerland, 2017; pp. 23–40. [Google Scholar]
- Bello, F.D.; Giannella, M.; Giorgioni, G.; Piergentili, A.; Quaglia, W. Receptor ligands as helping hands to L-DOPA in the treatment of Parkinson’s disease. Biomolecules 2019, 9, 142. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sadatsafavi, M.; Tavakoli, H.; Samii, A.; Etminan, M. Effects of β2-Adrenergic Agonists on Risk of Parkinson’s Disease in COPD: A Population-Based Study. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Ton, T.G.; Heckbert, S.R.; Longstreth, W., Jr.; Rossing, M.A.; Kukull, W.A.; Franklin, G.M.; Swanson, P.D.; Smith-Weller, T.; Checkoway, H. Calcium channel blockers and β-blockers in relation to Parkinson’s disease. Park. Relat. Disord. 2007, 13, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.; Ismond, K.P.; Chapman, E.; Flood, P. Potential benefits of therapeutic use of β2-adrenergic receptor agonists in neuroprotection and Parkinson’s disease. J. Immunol. Res. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Koren, G.; Norton, G.; Radinsky, K.; Shalev, V. Chronic Use of β-Blockers and the Risk of Parkinson’s Disease. Clin. Drug Investig. 2019, 39, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Gronich, N.; Abernethy, D.R.; Auriel, E.; Lavi, I.; Rennert, G.; Saliba, W. β2-adrenoceptor agonists and antagonists and risk of Parkinson’s disease. Mov. Disord. 2018, 33, 1465–1471. [Google Scholar] [CrossRef]
- Chen, C.-L.; Wang, S.-Y.; Chen, T.-C.; Chuang, C.-S. Association between β2-adrenoreceptor medications and risk of Parkinson’s disease: A meta-analysis. Medicina 2021, 57, 1006. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, E.; Yssel, J.D.; McNamara, C.; Harkin, A. Pharmacological targeting of β2-adrenoceptors is neuroprotective in the LPS inflammatory rat model of Parkinson’s disease. Br. J. Pharmacol. 2020, 177, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.P. Current issues with β2-adrenoceptor agonists: Pharmacology and molecular and cellular mechanisms. Clin. Rev. Allergy Immunol. 2006, 31, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Azevedo Voltarelli, V.; Coronado, M.; Gonçalves Fernandes, L.; Cruz Campos, J.; Jannig, P.R.; Batista Ferreira, J.C.; Fajardo, G.; Chakur Brum, P.; Bernstein, D. β2-Adrenergic signaling modulates mitochondrial function and morphology in skeletal muscle in response to aerobic exercise. Cells 2021, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Ge, M.-M.; Li, D.-Y.; Wang, X.-M.; Liu, D.-Q.; Ye, D.-W.; Tian, Y.-K.; Zhou, Y.-Q.; Chen, J.-P. β2-adrenoreceptor agonist ameliorates mechanical allodynia in paclitaxel-induced neuropathic pain via induction of mitochondrial biogenesis. Biomed. Pharmacother. 2021, 144, 112331. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.R.; Zhang, X.N.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-H.; Nakamura, T.; Lipton, S.A. Mitochondrial dynamics in cell death and neurodegeneration. Cell. Mol. Life Sci. 2010, 67, 3435–3447. [Google Scholar] [CrossRef]
- Lin, C.-H.; Tsai, P.-I.; Lin, H.-Y.; Hattori, N.; Funayama, M.; Jeon, B.; Sato, K.; Abe, K.; Mukai, Y.; Takahashi, Y.; et al. Mitochondrial UQCRC1 mutations cause autosomal dominant parkinsonism with polyneuropathy. Brain 2020, 143, 3352–3373. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, P.L.; Tai, C.H.; Lin, H.I.; Chen, C.S.; Chen, M.L.; Wu, R.M. A clinical and genetic study of early-onset and familial parkinsonism in Taiwan: An integrated approach combining gene dosage analysis and next-generation sequencing. Mov. Disord. 2019, 34, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.-C.; Huang, K.-L.; Chen, P.-L.; Li, J.-L.; Lu, S.H.-A.; Chang, J.-C.; Lin, H.-Y.; Lo, W.-C.; Huang, S.-Y.; Lee, T.-T.; et al. UQCRC1 engages cytochrome c for neuronal apoptotic cell death. Cell Rep. 2021, 36, 109729. [Google Scholar] [CrossRef]
- Li, J.-L.; Lin, T.-Y.; Chen, P.-L.; Guo, T.-N.; Huang, S.-Y.; Chen, C.-H.; Lin, C.-H.; Chan, C.-C. Mitochondrial function and Parkinson’s disease: From the perspective of the electron transport chain. Front. Mol. Neurosci. 2021, 14, 797833. [Google Scholar] [CrossRef]
- Chang, J.-C.; Wu, S.-L.; Liu, K.-H.; Chen, Y.-H.; Chuang, C.-S.; Cheng, F.-C.; Su, H.-L.; Wei, Y.-H.; Kuo, S.-J.; Liu, C.-S. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: Restoration of mitochondria functions and attenuation of 6-hydroxydopamine–induced neurotoxicity. Transl. Res. 2016, 170, 40–56.e3. [Google Scholar] [CrossRef]
- Peng, J.-Y.; Lin, C.-C.; Chen, Y.-J.; Kao, L.-S.; Liu, Y.-C.; Chou, C.-C.; Huang, Y.-H.; Chang, F.-R.; Wu, Y.-C.; Tsai, Y.-S.; et al. Automatic morphological subtyping reveals new roles of caspases in mitochondrial dynamics. PLoS Comput. Biol. 2011, 7, e1002212. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-C.; Fan, X.; Hu, C.-H.; Chao, Y.-C.; Liu, C.-S.; Chang, J.-C.; Sen, Y. Comparison of mitochondrial transplantation by using a stamp-type multineedle injector and platelet-rich plasma therapy for hair aging in naturally aging mice. Biomed. Pharmacother. 2020, 130, 110520. [Google Scholar] [CrossRef] [PubMed]
- Suh, B.K.; Lee, S.; Park, C.; Suh, Y.; Kim, S.J.; Woo, Y.; Nhung, T.T.M.; Lee, S.B.; Mun, D.J.; Goo, B.S.; et al. Schizophrenia-associated dysbindin modulates axonal mitochondrial movement in cooperation with p150glued. Mol. Brain 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Venkatesh, T. YBX1/lncRNA SBF2-AS1 interaction regulates proliferation and tamoxifen sensitivity via PI3K/AKT/MTOR signaling in breast cancer cells. Mol. Biol. Rep. 2023, 50, 3413–3428. [Google Scholar] [CrossRef] [PubMed]
- Vekaria, H.J.; Hubbard, W.B.; Scholpa, N.E.; Spry, M.L.; Gooch, J.L.; Prince, S.J.; Schnellmann, R.G.; Sullivan, P.G. Formoterol, a β2-adrenoreceptor agonist, induces mitochondrial biogenesis and promotes cognitive recovery after traumatic brain injury. Neurobiol. Dis. 2020, 140, 104866. [Google Scholar] [CrossRef] [PubMed]
- Cameron, R.B.; Gibbs, W.S.; Miller, S.R.; Dupre, T.V.; Megyesi, J.; Beeson, C.C.; Schnellmann, R.G. Proximal tubule β2-adrenergic receptor mediates formoterol-induced recovery of mitochondrial and renal function after ischemia-reperfusion injury. J. Pharmacol. Exp. Ther. 2019, 369, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, K.H.; Brosius, F.C., III; Schnellmann, R.G. Regulation of mitochondrial dynamics and energetics in the diabetic renal proximal tubule by the β2-adrenergic receptor agonist formoterol. Am. J. Physiol.-Ren. Physiol. 2020, 319, F773–F779. [Google Scholar] [CrossRef] [PubMed]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, J.; Ma, D.; Wang, F.; Wang, Y.; Li, C.; Wang, X.; Yin, X.; Zhang, M.; Dagda, R.K.; et al. Neuroprotective mitochondrial remodeling by AKAP121/PKA protects HT22 cell from glutamate-induced oxidative stress. Mol. Neurobiol. 2019, 56, 5586–5607. [Google Scholar] [CrossRef] [PubMed]
- van der Stel, W.; Yang, H.; le Dévédec, S.E.; van de Water, B.; Beltman, J.B.; Danen, E.H. High-content high-throughput imaging reveals distinct connections between mitochondrial morphology and functionality for OXPHOS complex I, III, and V inhibitors. Cell Biol. Toxicol. 2022, 39, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Chen, S.; Xiong, D.; Wang, H.; Zhu, L.; Wei, Y.; Li, Y.; Zou, S. Mitochondrial Dynamics: Working with the Cytoskeleton and Intracellular Organelles to Mediate Mechanotransduction. Aging Dis. 2023, 14, 1511–1532. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Matsuki, N.; Koyama, R. AMP-activated protein kinase mediates activity-dependent axon branching by recruiting mitochondria to axon. Dev. Neurobiol. 2014, 74, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Burska, D.; Stiburek, L.; Krizova, J.; Vanisova, M.; Martinek, V.; Sladkova, J.; Zamecnik, J.; Honzik, T.; Zeman, J.; Hansikova, H.; et al. Homozygous missense mutation in UQCRC2 associated with severe encephalomyopathy, mitochondrial complex III assembly defect and activation of mitochondrial protein quality control. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166147. [Google Scholar] [CrossRef]
- El Fissi, N.; Rojo, M.; Aouane, A.; Karatas, E.; Poliacikova, G.; David, C.; Royet, J.; Rival, T. Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT 2A neuropathy. EMBO Rep. 2018, 19, e45241. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.-G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial homeostasis and cellular senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Kim, T.; Jeon, J.; Jang, Y.; Kim, P.B.; Lopes, C.; Leblanc, P.; Cohen, B.M.; Kim, K.-S. SIRT2 regulates mitochondrial dynamics and reprogramming via MEK1-ERK-DRP1 and AKT1-DRP1 axes. Cell Rep. 2021, 37, 110155. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Chen, X.; Chen, X.; Li, J.; Cheng, B.; Xia, J. Mitochondrial dynamics: Fission and fusion in fate determination of mesenchymal stem cells. Front. Cell Dev. Biol. 2020, 8, 580070. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.; Cui, C.; Huang, X.; Yin, X.; Li, Y.; Wen, J.; Luan, Q. MFN2 silencing promotes neural differentiation of embryonic stem cells via the Akt signaling pathway. J. Cell. Physiol. 2020, 235, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Gasser, T. Update on the genetics of Parkinson’s disease. Mov. Disord. 2007, 22, S343–S350. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Charry, L.; Cookson, M.R.; Niño, A.; Arboleda, H.; Arboleda, G. Downregulation of Pink1 influences mitochondrial fusion–fission machinery and sensitizes to neurotoxins in dopaminergic cells. Neurotoxicology 2014, 44, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK 1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guardia-Laguarta, C.; Yin, J.; Erdjument-Bromage, H.; Martin, B.; James, M.; Jiang, X.; Przedborski, S. The ubiquitination of PINK1 is restricted to its mature 52-kDa form. Cell Rep. 2017, 20, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Fedorowicz, M.A.; de Vries-Schneider, R.L.; Rüb, C.; Becker, D.; Huang, Y.; Zhou, C.; Alessi Wolken, D.M.; Voos, W.; Liu, Y.; Przedborski, S. Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 2014, 15, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Soman, S.K.; Dagda, R.K. Role of Cleaved PINK1 in Neuronal Development, Synaptogenesis, and Plasticity: Implications for Parkinson’s Disease. Front. Neurosci. 2021, 15, 769331. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, P.; Huang, R.; Wang, C.; Sun, L.; Lan, B.; He, Y.; Zhao, H.; Gao, Y. PINK1: The guard of mitochondria. Life Sci. 2020, 259, 118247. [Google Scholar] [CrossRef] [PubMed]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. npj Regen. Med. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.-H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Drerup, C.M. Axonal transport and mitochondrial function in neurons. Front. Cell. Neurosci. 2019, 13, 373. [Google Scholar] [CrossRef] [PubMed]
- Ishida, A.; Ohta, T.; Toda, A.; Murata, K.; Usui, C.; Narita, N.; Tanaka, T.; Otani, H. Formoterol, a Long-Acting Beta2 Adrenergic Agonist, Improves Cognitive Function and Promotes Dendritic Complexity in a Mouse Model of Down Syndrome. J. Alzheimer’s Dis. 2013, 37, 557–570. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, J.-C.; Chang, H.-S.; Chao, Y.-C.; Huang, C.-S.; Lin, C.-H.; Wu, Z.-S.; Chang, H.-J.; Liu, C.-S.; Chuang, C.-S. Formoterol Acting via β2-Adrenoreceptor Restores Mitochondrial Dysfunction Caused by Parkinson’s Disease-Related UQCRC1 Mutation and Improves Mitochondrial Homeostasis Including Dynamic and Transport. Biology 2024, 13, 231. https://doi.org/10.3390/biology13040231
Chang J-C, Chang H-S, Chao Y-C, Huang C-S, Lin C-H, Wu Z-S, Chang H-J, Liu C-S, Chuang C-S. Formoterol Acting via β2-Adrenoreceptor Restores Mitochondrial Dysfunction Caused by Parkinson’s Disease-Related UQCRC1 Mutation and Improves Mitochondrial Homeostasis Including Dynamic and Transport. Biology. 2024; 13(4):231. https://doi.org/10.3390/biology13040231
Chicago/Turabian StyleChang, Jui-Chih, Huei-Shin Chang, Yi-Chun Chao, Ching-Shan Huang, Chin-Hsien Lin, Zhong-Sheng Wu, Hui-Ju Chang, Chin-San Liu, and Chieh-Sen Chuang. 2024. "Formoterol Acting via β2-Adrenoreceptor Restores Mitochondrial Dysfunction Caused by Parkinson’s Disease-Related UQCRC1 Mutation and Improves Mitochondrial Homeostasis Including Dynamic and Transport" Biology 13, no. 4: 231. https://doi.org/10.3390/biology13040231
APA StyleChang, J.-C., Chang, H.-S., Chao, Y.-C., Huang, C.-S., Lin, C.-H., Wu, Z.-S., Chang, H.-J., Liu, C.-S., & Chuang, C.-S. (2024). Formoterol Acting via β2-Adrenoreceptor Restores Mitochondrial Dysfunction Caused by Parkinson’s Disease-Related UQCRC1 Mutation and Improves Mitochondrial Homeostasis Including Dynamic and Transport. Biology, 13(4), 231. https://doi.org/10.3390/biology13040231