
Double-Edged Sword: Exploring the Mitochondria–Complement Bidirectional Connection in Cellular Response and Disease




Abstract
Simple Summary
Abstract
1. Introduction
2. Mitochondria
3. Complement System
3.1. Internalization Mechanisms of Complement Proteins
3.2. Intracellular Complement System
3.3. Interaction of Complosome with Mitochondria
4. Role of Mitochondrial DAMPs in Complement Response
4.1. Intracellular Interactions and Impacts
Cytochrome C, Apoptosis, and Complement System
4.2. Extracellular Interactions and Impacts
Cardiolipin and Complement System
4.3. Intracellular and Extracellular Interactions and Impacts
4.3.1. ATP and Complement System
4.3.2. ROS and Complement System
4.4. Complement System Damages Mitochondria: A Vicious Cycle

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial DAMPs | Interactions with Complement | Reference |
|---|---|---|
| Cytochrome c | C3 | [54] |
| C5a | [55,56] | |
| Cardiolipin | C1 | [57,58] |
| ATP | MBL | [59,60] |
| ROS | iC3b | [17] |
| MBL | [63] | |
| C5a | [69] |
5. Interplay of Complement and Mitochondria in Human Diseases
5.1. Alzheimer’s Disease
5.2. Age-Related Macular Degeneration
5.3. Myocardial Ischemia
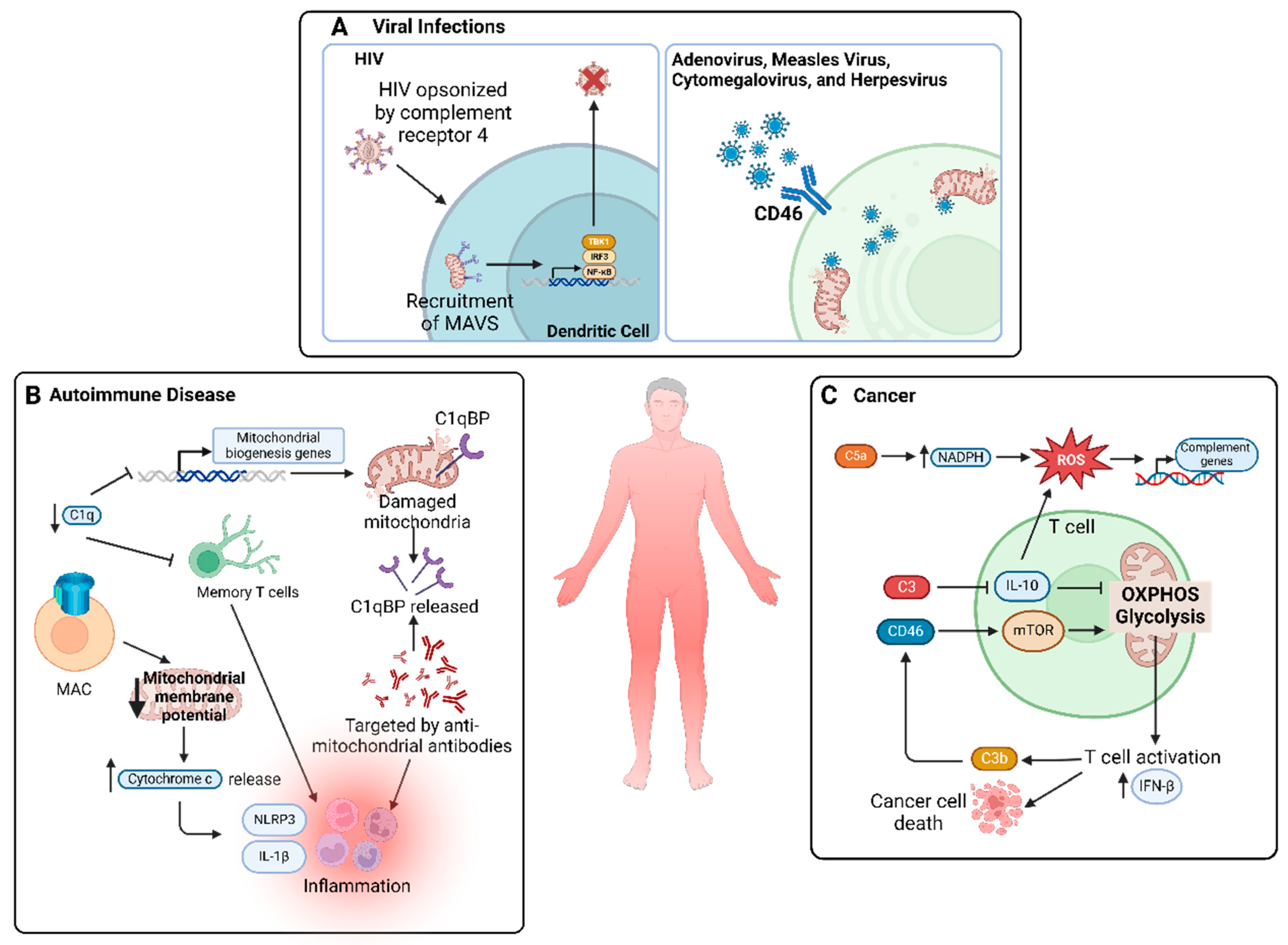
5.4. Viral Infections
5.5. Autoimmune Diseases
5.6. Cancer
6. Therapeutic Potential of Targeting Mitochondria and Complement System in Disease Intervention
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OXPHOS | Oxidative phosphorylation |
| DAMP | Damage-associated molecular pattern |
| ROS | Reactive oxidative species |
| mtDNA | Mitochondrial DNA |
| IFN | Interferon |
| C3 | Complement component 3 |
| C5 | Complement component 5 |
| C1q | Complement component 1q |
| iC3b | Inactivated C3b |
| MAC | Membrane attack complex |
| GPI | Glycosylphosphatidylinositol |
| C3aR | C3a receptor |
| C5aR | C5a receptor |
| CFH | Complement factor H |
| CTSL | Cathepsin L |
| MAVS | Mitochondrial antiviral signaling |
| IL | Interleukin |
| C1qBP | C1q-binding protein |
| I/R | Ischemia/reperfusion |
| NLRP3 | NOD-, LRR-, and pyrin domain-containing protein 3 |
| NF-κB | Nuclear factor kappa B |
| MBL | Mannose-binding lectin |
| AD | Alzheimer’s disease |
| Aβ | Amyloid beta |
| AMD | Age-related macular degeneration |
| MI | Myocardial infarction |
| HIV | Human immunodeficiency virus |
| SLE | Systemic lupus erythematosus |
| HSP | Heat shock protein |
References
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef]
- Mohamud, Y.; Li, B.Z.; Bahreyni, A.; Luo, H.L. Mitochondria dysfunction at the heart of viral myocarditis: Mechanistic insights and therapeutic implications. Viruses 2023, 15, 351. [Google Scholar] [CrossRef]
- Guan, P.P.; Ge, T.Q.; Wang, P. As a potential therapeutic target, C1q induces synapse loss via inflammasome-activating apoptotic and mitochondria impairment mechanisms in Alzheimer’s disease. J. Neuroimmune Pharm. 2023, 18, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Lo, M.R.W.; Woodruff, T.M. Complement: Bridging the innate and adaptive immune systems in sterile inflammation. J. Leukoc. Biol. 2020, 108, 339–351. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Le Friec, G.; Kemper, C. Complement—tapping into new sites and effector systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Rahman, J.; Singh, P.; Merle, N.S.; Niyonzima, N.; Kemper, C. Complement’s favourite organelle-Mitochondria? Brit J. Pharmacol. 2021, 178, 2771–2785. [Google Scholar] [CrossRef]
- West, E.E.; Kemper, C. Complosome—The intracellular complement system. Nat. Rev. Nephrol. 2023, 19, 426–439. [Google Scholar] [CrossRef]
- Kolev, M.; Dimeloe, S.; Le Friec, G.; Navarini, A.; Arbore, G.; Povoleri, G.A.; Fischer, M.; Belle, R.; Loeliger, J.; Develioglu, L.; et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity 2015, 42, 1033–1047. [Google Scholar] [CrossRef]
- Hess, C.; Kemper, C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity 2016, 45, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef] [PubMed]
- Reis, E.S.; Mastellos, D.C.; Hajishengallis, G.; Lambris, J.D. New insights into the immune functions of complement. Nat. Rev. Immunol. 2019, 19, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Wu, M.H. Pattern recognition receptors in health and diseases. Signal Transduct. Tar. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Kagan, J.C. Activation and pathogenic manipulation of the sensors of the innate immune system. Microbes Infect. 2017, 19, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Vakeva, A.; Bukusoglu, C.; Zund, G.; Sperati, C.J.; Colgan, S.P.; Stahl, G.L. Reoxygenation of hypoxic human umbilical vein endothelial cells activates the classic complement pathway. Circulation 1997, 96, 326–333. [Google Scholar] [CrossRef]
- Collard, C.D.; Agah, A.; Stahl, G.L. Complement activation following reoxygenation of hypoxic human endothelial cells: Role of intracellular reactive oxygen species, NF-κB and new protein synthesis. Immunopharmacology 1998, 39, 39–50. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Ryan, M.T.; Hoogenraad, N.J. Mitochondrial-nuclear communications. Annu. Rev. Biochem. 2007, 76, 701–722. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Mohamud, Y.; Xue, Y.C.; Liu, H.T.; Ng, C.S.; Bahreyni, A.; Luo, H.L. Autophagy receptor protein Tax1-binding protein 1/TRAF6-binding protein is a cellular substrate of enteroviral proteinase. Front. Microbiol. 2021, 12, 647410. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef]
- Mohamud, Y.; Shi, J.; Tang, H.; Xiang, P.; Xue, Y.C.; Liu, H.; Ng, C.S.; Luo, H. Coxsackievirus infection induces a non-canonical autophagy independent of the ULK and PI3K complexes. Sci. Rep. 2020, 10, 19068. [Google Scholar] [CrossRef]
- Nakahira, K.; Hisata, S.; Choi, A.M.K. The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid. Redox Sign 2015, 23, 1329–1350. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.M.; Kalpage, H.A.; Vaishnav, A.; Liu, J.; Lee, I.; Mahapatra, G.; Turner, A.A.; Zurek, M.P.; Ji, Q.Q.; Moraes, C.T.; et al. Regulation of respiration and apoptosis by cytochrome c threonine 58 phosphorylation. Sci. Rep. 2019, 9, 15815. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Mohamud, Y.; Fu, C.; Fan, Y.M.; Zhang, Y.L.; Lin, J.F.C.; Hwang, S.W.; Wang, Z.C.; Luo, H. Activation of cGAS-STING suppresses coxsackievirus replication via interferon-dependent signaling. Antivir. Res. 2024, 222, 105811. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. The complement system and innate immunity. In Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- King, B.C.; Kulak, K.; Krus, U.; Rosberg, R.; Golec, E.; Wozniak, K.; Gomez, M.F.; Zhang, E.M.; O’Connell, D.J.; Renström, E.; et al. Complement component C3 is highly expressed in human pancreatic islets and prevents β cell death via ATG16L1 interaction and autophagy regulation. Cell Metab. 2019, 29, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Kremlitzka, M.; Colineau, L.; Nowacka, A.A.; Mohlin, F.C.; Wozniak, K.; Blom, A.M.; King, B. Alternative translation and retrotranslocation of cytosolic C3 that detects cytoinvasive bacteria. Cell Mol. Life Sci. 2022, 79, 291. [Google Scholar] [CrossRef] [PubMed]
- Golec, E.; Rosberg, R.; Zhang, E.M.; Renström, E.; Blom, A.M.; King, B. A cryptic non-GPI-anchored cytosolic isoform of CD59 controls insulin exocytosis in pancreatic β-cells by interaction with SNARE proteins. FASEB J. 2019, 33, 12425–12434. [Google Scholar] [CrossRef] [PubMed]
- Strainic, M.G.; Liu, J.B.; Huang, D.P.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.; Heeger, P.S.; Medof, M.E. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4 T cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Le Friec, G.; Kemper, C. The role of complement in CD4 T cell homeostasis and effector functions. Semin. Immunol. 2013, 25, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Lubbers, R.; van Essen, M.F.; van Kooten, C.; Trouw, L.A. Production of complement components by cells of the immune system. Clin. Exp. Immunol. 2017, 188, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; Gasque, P. Extrahepatic complement biosynthesis: Where, when and why? Clin. Exp. Immunol. 1997, 107, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tenner, A.J.; Volkin, D.B. Complement subcomponent-C1q secreted by cultured human-monocytes has subunit structure identical with that of serum-C1q. Biochem. J. 1986, 233, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Beeson, G.; Beeson, C.; Rohrer, B. Mitochondrial C3a receptor activation in oxidatively stressed epithelial cells reduces mitochondrial respiration and metabolism. Front. Immunol. 2021, 12, 628062. [Google Scholar] [CrossRef] [PubMed]
- Arbore, G.; West, E.E.; Spolski, R.; Robertson, A.A.B.; Klos, A.; Rheinheimer, C.; Dutow, P.; Woodruff, T.M.; Yu, Z.X.; O’Neill, L.A.; et al. T Helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4 T Cells. Science 2016, 352, aad1210. [Google Scholar] [CrossRef]
- Martin, M.; Leffler, J.; Smolag, K.I.; Mytych, J.; Björk, A.; Chaves, L.D.; Alexander, J.J.; Quigg, R.J.; Blom, A.M. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Differ. 2016, 23, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.C.H.; Bidgood, S.R.; McEwan, W.A.; James, L.C. Intracellular sensing of complement C3 activates cell autonomous immunity. Science 2014, 345, 1256070. [Google Scholar] [CrossRef] [PubMed]
- Sorbara, M.T.; Foerster, E.G.; Tsalikis, J.; Abdel-Nour, M.; Mangiapane, J.; Sirluck-Schroeder, I.; Tattoli, I.; van Dalen, R.; Isenman, D.E.; Rohde, J.R.; et al. Complement C3 drives autophagy-dependent restriction of cyto-invasive bacteria. Cell Host Microbe 2018, 23, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Niyonzima, N.; Rahman, J.; Kunz, N.; West, E.E.; Freiwald, T.; Desai, J.; Merle, N.S.; Gidon, A.; Sporsheim, B.; Lionakis, M.S.; et al. Mitochondrial C5aR1 activity in macrophages controls IL-1β production underlying sterile inflammation. Sci. Immunol. 2021, 6, eabf2489. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Xu, Y.; Li, L.; Lv, X.; Li, L.; Chen, J.; Zhou, D.; Wang, X.; Wang, Q.; Zhang, W.; et al. Intracellular complement C5a/C5aR1 stabilizes beta-catenin to promote colorectal tumorigenesis. Cell Rep. 2022, 39, 110851. [Google Scholar] [CrossRef] [PubMed]
- Daugan, M.V.; Revel, M.; Russick, J.; Dragon-Durey, M.A.; Gaboriaud, C.; Robe-Rybkine, T.; Poillerat, V.; Grunenwald, A.; Lacroix, G.; Bougouin, A.; et al. Complement C1s and C4d as prognostic biomarkers in renal cancer: Emergence of noncanonical functions of C1s. Cancer Immunol. Res. 2021, 9, 891–908. [Google Scholar] [CrossRef] [PubMed]
- West, E.E.; Kolev, M.; Kemper, C. Complement and the regulation of T cell responses. Annu. Rev. Immunol. 2018, 36, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.Z.; Zhang, Y.; Krainer, A.R.; Xu, R.M. Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc. Natl. Acad. Sci. USA 1999, 96, 3572–3577. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. A mitochondrial checkpoint in autoimmune disease. Cell Metab. 2018, 28, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.R.; Ghosh, O.; Datta, K. Excessive reactive oxygen species induces apoptosis in fibroblasts: Role of mitochondrially accumulated hyaluronic acid binding protein 1 (HABP1/p32/gC1qR). Exp. Cell Res. 2008, 314, 651–667. [Google Scholar] [CrossRef]
- Galvan, M.D.; Greenlee-Wacker, M.C.; Bohlson, S.S. C1q and phagocytosis: The perfect complement to a good meal. J. Leukoc. Biol. 2012, 92, 489–497. [Google Scholar] [CrossRef]
- McGee, A.M.; Douglas, D.L.; Liang, Y.Y.; Hyder, S.M.; Baines, C.P. The mitochondrial protein C1qbp promotes cell proliferation, migration and resistance to cell death. Cell Cycle 2011, 10, 4119–4127. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Lee, H.K.Y.; Liu, J.Y.; Wong, K.A.; Brown, L.M.; Li, X.; Xiaoli, A.M.; Yang, F.J.; Zhang, M. Complement C3 reduces apoptosis via interaction with the intrinsic apoptotic pathway. Cells 2023, 12, 2282. [Google Scholar] [CrossRef]
- De Hoog, V.C.; Timmers, L.; Van Duijvenvoorde, A.; De Jager, S.C.A.; Van Middelaar, B.J.; Smeets, M.B.; Woodruff, T.M.; Doevendans, P.A.; Pasterkamp, G.; Hack, C.E.; et al. Leucocyte expression of complement C5a receptors exacerbates infarct size after myocardial reperfusion injury. Cardiovasc. Res. 2014, 103, 521–529. [Google Scholar] [CrossRef]
- Vakeva, A.P.; Agah, A.; Rollins, S.A.; Matis, L.A.; Li, L.; Stahl, G.L. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion—Role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 1998, 97, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Rossen, R.D.; Michael, L.H.; Hawkins, H.K.; Youker, K.; Dreyer, W.J.; Baughn, R.E.; Entman, M.L. Cardiolipin-protein complexes and initiation of complement activation after coronary artery occlusion. Circ. Res. 1994, 75, 546–555. [Google Scholar] [CrossRef]
- Peitsch, M.C.; Tschopp, J.; Kress, A.; Isliker, H. Antibody-independent activation of the complement-system by mitochondria is mediated by cardiolipin. Biochem. J. 1988, 249, 495–500. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Thapa, A.; Bujko, K.; Brzezniakiewicz-Janus, K.; Lenkiewicz, A.M. NLRP3 inflammasome couples purinergic signaling with activation of the complement cascade for the optimal release of cells from bone marrow. Leukemia 2019, 33, 815–825. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Adamiak, M.; Plonka, M.; Abdel-Latif, A.; Ratajczak, J. Mobilization of hematopoietic stem cells as a result of innate immunity-mediated sterile inflammation in the bone marrow microenvironment-the involvement of extracellular nucleotides and purinergic signaling. Leukemia 2018, 32, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Mack, A.; Bujko, K.; Domingues, A.; Pedziwiatr, D.; Kucia, M.; Ratajczak, J.; Ulrich, H.; Kucharska-Mazur, J.; Samochowiec, J. ATP-Nlrp3 inflammasome-complement cascade axis in sterile brain inflammation in psychiatric patients and its impact on stem cell trafficking. Stem Cell Rev. Rep. 2019, 15, 497–505. [Google Scholar] [CrossRef]
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Sign 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed]
- Collard, C.D.; Väkevä, A.; Morrissey, M.A.; Agah, A.; Rollins, S.A.; Reenstra, W.R.; Buras, J.A.; Meri, S.; Stahl, G.L. Complement activation after oxidative stress: Role of the lectin complement pathway. Am. J. Pathol. 2000, 156, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.S.; Jeon, H.; Yoo, S.M.; Lee, M.S. Activation of the complement system on human endothelial cells by urban particulate matter triggers inflammation-related protein production. Int. J. Mol. Sci. 2021, 22, 3336. [Google Scholar] [CrossRef] [PubMed]
- Villalpando-Rodriguez, G.E.; Gibson, S.B. Reactive oxygen species (ROS) regulates different types of cell death by acting as a rheostat. Oxid. Med. Cell Longev. 2021, 2021, 9912436. [Google Scholar] [CrossRef] [PubMed]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern recognition receptors and the host cell death molecular machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed]
- Trakkides, T.O.; Schäfer, N.; Reichenthaler, M.; Kühn, K.; Brandwijk, R.J.M.G.E.; Toonen, E.J.M.; Urban, F.; Wegener, J.; Enzmann, V.; Pauly, D. Oxidative stress increases endogenous complement-dependent inflammatory and angiogenic responses in retinal pigment epithelial cells independently of exogenous complement sources. Antioxidants 2019, 8, 548. [Google Scholar] [CrossRef]
- Nord, D.M. Activated Complement Products Contribute to Mitochondrial Damage of the Airway Epithelium during Lung Transplantation; Medical University of South Carolina: Charleston, SC, USA, 2020. [Google Scholar]
- Tsai, I.J.; Lin, W.C.; Yang, Y.H.; Tseng, Y.L.; Lin, Y.H.; Chou, C.H.; Tsau, Y.K. High Concentration of C5a-Induced Mitochondria-Dependent Apoptosis in Murine Kidney Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 4465. [Google Scholar] [CrossRef]
- Crivelli, S.M.; Quadri, Z.; Vekaria, H.J.; Zhu, Z.H.; Tripathi, P.; Elsherbini, A.; Zhang, L.P.; Sullivan, P.G.; Bieberich, E. Inhibition of acid sphingomyelinase reduces reactive astrocyte secretion of mitotoxic extracellular vesicles and improves Alzheimer’s disease pathology in the 5xFAD mouse. Acta Neuropathol. Com. 2023, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Luo, X.G.; Weber, G.A.; Zheng, J.L.; Gendelman, H.E.; Ikezu, T. C1q-calreticulin induced oxidative neurotoxicity: Relevance for the neuropathogenesis of Alzheimer’s disease. J. Neuroimmunol. 2003, 135, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef] [PubMed]
- Datta, D.; Leslie, S.N.; Morozov, Y.M.; Duque, A.; Rakic, P.; van Dyck, C.H.; Nairn, A.C.; Arnsten, A.F.T. Classical complement cascade initiating C1q protein within neurons in the aged rhesus macaque dorsolateral prefrontal cortex. J. Neuroinflamm 2020, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Holubowicz, R.; Wojtas, M.; Taube, M.; Kozak, M.; Ozyhar, A.; Dobryszycki, P. Effect of calcium ions on structure and stability of the C1q-like domain of otolin-1 from human and zebrafish. FEBS J. 2017, 284, 4278–4297. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jo, J.; Jia, J.M.; Lo, S.C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Vyawahare, H.; Shinde, P. Age-related macular degeneration: Epidemiology, pathophysiology, diagnosis, and treatment. Cureus J. Med. Sci. 2022, 14, e29583. [Google Scholar] [CrossRef]
- Kenney, M.C.; Chwa, M.; Atilano, S.R.; Pavlis, J.M.; Falatoonzadeh, P.; Ramirez, C.; Malik, D.; Hsu, T.; Woo, G.; Soe, K.; et al. Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: Implications for age-related macular degeneration. PLoS ONE 2013, 8, e54339. [Google Scholar] [CrossRef]
- Juel, H.B.; Kaestel, C.; Folkersen, L.; Faber, C.; Heegaard, N.H.H.; Borup, R.; Nissen, M.H. Retinal pigment epithelial cells upregulate expression of complement factors after co-culture with activated T cells. Exp. Eye Res. 2011, 92, 180–188. [Google Scholar] [CrossRef]
- Thurman, J.M.; Renner, B.; Kunchithapautham, K.; Ferreira, V.P.; Pangburn, M.K.; Ablonczy, Z.; Tomlinson, S.; Holers, V.M.; Rohrer, B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 2009, 284, 16939–16947. [Google Scholar] [CrossRef]
- Bellizzi, D.; Cavalcante, P.; Taverna, D.; Rose, G.; Passarino, G.; Salvioli, S.; Franceschi, C.; De Benedictis, G. Gene expression of cytokines and cytokine receptors is modulated by the common variability of the mitochondrial DNA in cybrid cell lines. Genes. Cells 2006, 11, 883–891. [Google Scholar] [CrossRef]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Köhl, J.; Cook, H.T.; Kemper, C. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 2013, 122, 3473–3481. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Moraes-Silva, I.C.; Rodrigues, B.; Coelho, H.J.; Feriani, D.J.; Irigoyen, M.C. Myocardial infarction and exercise training: Evidence from basic science. Adv. Exp. Med. Biol. 2017, 999, 139–153. [Google Scholar] [CrossRef]
- Salari, N.; Morddarvanjoghi, F.; Abdolmaleki, A.; Rasoulpoor, S.; Khaleghi, A.A.; Hezarkhani, L.A.; Shohaimi, S.; Mohammadi, M. The global prevalence of myocardial infarction: A systematic review and meta-analysis. BMC Cardiovasc. Disor. 2023, 23. [Google Scholar] [CrossRef]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: The insight into mechanisms and therapeutic potentials. Brit J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef]
- Pinckard, R.N.; Olson, M.S.; Giclas, P.C.; Terry, R.; Boyer, J.T.; O’Rourke, R.A. Consumption of classical complement components by heart subcellular membranes in vitro and in patients after acute myocardial infarction. J. Clin. Investig. 1975, 56, 740–750. [Google Scholar] [CrossRef]
- Kagiyama, A.; Savage, H.E.; Michael, L.H.; Hanson, G.; Entman, M.L.; Rossen, R.D. Molecular-basis of complement activation in ischemic myocardium—Identification of specific molecules of mitochondrial origin that bind human C1q and fix complement. Circ. Res. 1989, 64, 607–615. [Google Scholar] [CrossRef]
- Rossen, R.D.; Michael, L.H.; Kagiyama, A.; Savage, H.E.; Hanson, G.; Reisberg, M.A.; Moake, J.N.; Kim, S.H.; Self, D.; Weakley, S. Mechanism of complement activation after coronary artery occlusion: Evidence that myocardial ischemia in dogs causes release of constituents of myocardial subcellular origin that complex with human C1q in vivo. Circ. Res. 1988, 62, 572–584. [Google Scholar] [CrossRef]
- Rossen, R.D.; Swain, J.L.; Michael, L.H.; Weakley, S.; Giannini, E.; Entman, M.L. Selective accumulation of the first component of complement and leukocytes in ischemic canine heart muscle. A possible initiator of an extra myocardial mechanism of ischemic injury. Circ. Res. 1985, 57, 119–130. [Google Scholar] [CrossRef]
- Torp, M.K.; Ranheim, T.; Schjalm, C.; Hjorth, M.; Heiestad, C.M.; Dalen, K.T.; Nilsson, P.H.; Mollnes, T.E.; Pischke, S.E.; Lien, E.; et al. Intracellular complement component 3 attenuated ischemia-reperfusion injury in the isolated buffer-perfused mouse heart and is associated with improved metabolic homeostasis. Front. Immunol. 2022, 13, 870811. [Google Scholar] [CrossRef]
- Orrem, H.L.; Nilsson, P.H.; Pischke, S.E.; Grindheim, G.; Garred, P.; Seljeflot, I.; Husebye, T.; Aukrust, P.; Yndestad, A.; Andersen, G.O.; et al. Acute heart failure following myocardial infarction: Complement activation correlates with the severity of heart failure in patients developing cardiogenic shock. Esc. Heart Fail. 2018, 5, 292–301. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Tian, X.B.; Chen, J.J.; Wang, X.; Xie, Y.W.; Zhang, X.D.; Han, D.T.; Fu, H.J.; Yin, W.P.; Wu, N.P. Global, regional, and national HIV/AIDS disease burden levels and trends in 1990–2019: A systematic analysis for the global burden of disease 2019 study. Front Public Health 2023, 11, 1068664. [Google Scholar] [CrossRef]
- Posch, W.; Bermejo-Jambrina, M.; Steger, M.; Witting, C.; Diem, G.; Hortnagl, P.; Hackl, H.; Lass-Florl, C.; Huber, L.A.; Geijtenbeek, T.B.H.; et al. Complement potentiates immune sensing of HIV-1 and early type I interferon responses. mBio 2021, 12, e0240821. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Atkinson, J.P. Membrane cofactor protein (MCP.; CD46): Deficiency states and pathogen connections. Curr. Opin. Immunol. 2021, 72, 126–134. [Google Scholar] [CrossRef]
- Yanagawa, B.; Spiller, O.B.; Proctor, D.G.; Choy, J.; Luo, H.L.; Zhang, H.F.M.; Suarez, A.; Yang, D.C.; McManus, B.M. Soluble recombinant coxsackievirus and adenovirus receptor abrogates coxsackievirus B3-mediated pancreatitis and myocarditis in mice. J. Infect. Dis. 2004, 189, 1431–1439. [Google Scholar] [CrossRef]
- Morrison, B.J.; Abu-Asab, M.; Morris, J.C.; Steel, J.C. Localization of human recombinant adenoviral vectors to the mitochondria following transduction of human cell lines. Acta Virol. 2019, 63, 111–116. [Google Scholar] [CrossRef]
- Sato, H.; Hoshi, M.; Ikeda, F.; Fujiyuki, T.; Yoneda, M.; Kai, C. Downregulation of mitochondrial biogenesis by virus infection triggers antiviral responses by cyclic GMP-AMP synthase. PLoS Pathog. 2021, 17, e1009841. [Google Scholar] [CrossRef]
- Combs, J.A.; Norton, E.B.; Saifudeen, Z.R.; Bentrup, K.H.Z.; Katakam, P.V.; Morris, C.A.; Myers, L.; Kaur, A.; Sullivan, D.E.; Zwezdaryk, K.J. Human cytomegalovirus alters host cell mitochondrial function during acute infection. J. Virol. 2020, 94, e01183-19. [Google Scholar] [CrossRef]
- Li, L.; Chi, J.; Zhou, F.; Guo, D.; Wang, F.; Liu, G.; Zhang, C.; Yao, K. Human herpesvirus 6A induces apoptosis of HSB-2 cells via a mitochondrion-related caspase pathway. J. Biomed. Res. 2010, 24, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef] [PubMed]
- Orbai, A.M.; Truedsson, L.; Sturfelt, G.; Nived, O.; Fang, H.; Alarcón, G.S.; Gordon, C.; Merrill, J.T.; Fortin, P.R.; Bruce, I.N.; et al. Anti-C1q antibodies in systemic lupus erythematosus. Lupus 2015, 24, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Lintner, K.E.; Wu, Y.L.; Yang, Y.; Spencer, C.H.; Hauptmann, G.; Hebert, L.A.; Atkinson, J.P.; Yu, C.Y. Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front. Immunol. 2016, 7, 36. [Google Scholar] [CrossRef]
- Atkinson, J.P.; Liszewski, M.K.; Yu, C.Y. Clinical aspects of the complement system in systemic lupus erythematosus. In Systemic Lupus Erythematosus; Academic Press: Cambridge, MA, USA, 2021; pp. 113–122. [Google Scholar]
- Hughes, J.; Nangaku, M.; Alpers, C.E.; Shankland, S.J.; Couser, W.G.; Johnson, R.J. C5b-9 membrane attack complex mediates endothelial cell apoptosis in experimental glomerulonephritis. Am. J. Physiol.-Ren. 2000, 278, F747–F757. [Google Scholar] [CrossRef] [PubMed]
- Oleesky, D.A.; Daniels, R.H.; Williams, B.D.; Amos, N.; Morgan, B.P. Terminal complement complexes and C1/C1 inhibitor complexes in rheumatoid-arthritis and other arthritic conditions. Clin. Exp. Immunol. 1991, 84, 250–255. [Google Scholar]
- Biró, É.; Nieuwland, R.; Tak, P.P.; Pronk, L.M.; Schaap, M.C.L.; Sturk, A.; Hack, C.E. Activated complement components and complement activator molecules on the surface of cell-derived microparticles in patients with rheumatoid arthritis and healthy individuals. Ann. Rheum. Dis. 2007, 66, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Makinde, V.A.; Senaldi, G.; Jawad, A.S.M.; Berry, H.; Vergani, D. Reflection of disease-activity in rheumatoid-arthritis by indexes of activation of the classical complement pathway. Ann. Rheum. Dis. 1989, 48, 302–306. [Google Scholar] [CrossRef]
- Mascaro, J.M.; Hausmann, G.; Herrero, C.; Grau, J.M.; Cid, M.C.; Palou, J.; Mascaro, J.M. Membrane attack complex deposits in cutaneous lesions of dermatomyositis. J. Investig. Dermatol. 1995, 104, 665. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Ceponis, A.; Meri, S.; Vuorikoski, A.; Kortekangas, P.; Sorsa, T.; Sukura, A.; Santavirta, S. Complement in acute and chronic arthritides: Assessment of C3c, C9, and protectin (CD59) in synovial membrane. Ann. Rheum. Dis. 1996, 55, 888–894. [Google Scholar] [CrossRef]
- Abu-Shakra, M.; Gladman, D.D.; Urowitz, M.B.; Farewell, V. Anticardiolipin antibodies in systemic lupus erythematosus: Clinical and laboratory correlations. Am. J. Med. 1995, 99, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Goldenberg, M.; Rabinovici, J.; Lidor, A.L.; Dulitzky, M.; Gilburd, B.; Shoenfeld, Y.; Schiff, E. Anti-cardiolipin antibodies in fetal blood and amniotic fluid derived from patients with the anti-phospholipid syndrome. Hum. Reprod. 2000, 15, 1170–1172. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.H.; Yan, Z.Y.; Yang, A.M. Mitochondria in innate immunity signaling and its therapeutic implications in autoimmune diseases. Front. Immunol. 2023, 14, 1160035. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Chen, Q.; Xia, Y.M. Oxidative stress contributes to inflammatory and cellular damage in systemic lupus erythematosus: Cellular markers and molecular mechanism. J. Inflamm. Res. 2023, 16, 453–465. [Google Scholar] [CrossRef]
- Yevgi, R.; Demir, R. Oxidative stress activity of fingolimod in multiple sclerosis. Clin. Neurol. Neurosur 2021, 202, 106500. [Google Scholar] [CrossRef] [PubMed]
- Tobore, T.O. Oxidative/Nitroxidative stress and multiple sclerosis. J. Mol. Neurosci. 2021, 71, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic. Biol. Med. 2018, 125, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, X.; Zou, H.; Li, M. Oxidative stress and treg and Th17 dysfunction in systemic lupus erythematosus. Oxid. Med. Cell Longev. 2016, 2016, 2526174. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.F.; Zhang, J.D.; Hong, S.Z.; Li, H.C.; Lu, L.; Xie, G.Q.; Luo, W.Q.; Du, Y.; Xie, Z.J.; Han, X.L.; et al. Oxidative stress-induced aberrant lipid metabolism is an important causal factor for dysfunction of immunocytes from patients with systemic lupus erythematosus. Free Radic. Biol. Med. 2021, 163, 210–219. [Google Scholar] [CrossRef]
- Perl, A.; Gergely, P.; Nagy, G.; Koncz, A.; Banki, K. Mitochondrial hyperpolarization: A checkpoint of T-cell life, death and autoimmunity. Trends Immunol. 2004, 25, 360–367. [Google Scholar] [CrossRef]
- Becker, Y.L.C.; Gagné, J.P.; Julien, A.S.; Lévesque, T.; Allaeys, I.; Gougeard, N.; Rubio, V.; Boisvert, F.M.; Jean, D.; Wagner, E.; et al. Identification of mitofusin 1 and complement component 1q subcomponent binding protein as mitochondrial targets in systemic lupus erythematosus. Arthritis Rheumatol. 2022, 74, 1193–1203. [Google Scholar] [CrossRef]
- Saha, P.; Chowdhury, A.R.; Dutta, S.; Chatterjee, S.; Ghosh, I.; Datta, K. Autophagic vacuolation induced by excess ROS generation in HABP1/p32/gC1qR overexpressing fibroblasts and its reversal by polymeric hyaluronan. PLoS ONE 2013, 8, e78131. [Google Scholar] [CrossRef]
- Jimenez-Duran, G.; Kozole, J.; Peltier-Heap, R.; Dickinson, E.R.; Kwiatkowski, C.R.; Zappacosta, F.; Annan, R.S.; Galwey, N.W.; Nichols, E.M.; Modis, L.K.; et al. Complement membrane attack complex is an immunometabolic regulator of NLRP3 activation and IL-18 secretion in human macrophages. Front. Immunol. 2022, 13, 918551. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Frade, R.; Rodrigues-Lima, F.; Huang, S.Y.; Xie, K.P.; Guillaume, N.; Bar-Eli, M. Procathepsin-L, a proteinase that cleaves human C3 (the third component of complement), confers high tumorigenic and metastatic properties to human melanoma cells. Cancer Res. 1998, 58, 2733–2736. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Liszewski, M.K.; Kemper, C. Complement in motion: The evolution of CD46 from a complement regulator to an orchestrator of normal cell physiology. J. Immunol. 2019, 203, 3–5. [Google Scholar] [CrossRef]
- Le Friec, G.; Sheppard, D.; Whiteman, P.; Karsten, C.M.; Shamoun, S.A.T.; Laing, A.; Bugeon, L.; Dallman, M.J.; Melchionna, T.; Chillakuri, C.; et al. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat. Immunol. 2012, 13, 1213–1221. [Google Scholar] [CrossRef]
- Cardone, J.; Le Friec, G.; Vantourout, P.; Roberts, A.; Fuchs, A.; Jackson, I.; Suddason, T.; Lord, G.; Atkinson, J.P.; Cope, A.; et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat. Immunol. 2010, 11, 862–871. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Abramczyk, H.; Brozek-Pluska, B.; Kopec, M. Double face of cytochrome in cancers by Raman imaging. Sci. Rep. 2022, 12, 2120. [Google Scholar] [CrossRef]
- Maeda, H.; Akaike, T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochem. Mosc. 1998, 63, 854–865. [Google Scholar]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Nathan, C.; Ding, A. SnapShot: Reactive oxygen intermediates (ROI). Cell 2010, 140, 951. [Google Scholar] [CrossRef]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef]
- Markiewski, M.M.; DeAngelis, R.A.; Benencia, F.; Ricklin-Lichtsteiner, S.K.; Koutoulaki, A.; Gerard, C.; Coukos, G.; Lambris, J.D. Modulation of the antitumor immune response by complement. Nat. Immunol. 2008, 9, 1225–1235. [Google Scholar] [CrossRef]
- Oshi, M.; Gandhi, S.; Yan, L.; Tokumaru, Y.; Wu, R.R.; Yamada, A.; Matsuyama, R.; Endo, I.; Takabe, K. Abundance of reactive oxygen species (ROS) is associated with tumor aggressiveness, immune response, and worse survival in breast cancer. Breast Cancer Res. Tr. 2022, 194, 231–241. [Google Scholar] [CrossRef]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial Homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Rozenberg, P.; Ziporen, L.; Gancz, D.; Saar-Ray, M.; Fishelson, Z. Cooperation between Hsp90 and mortalin/GRP75 in resistance to cell death induced by complement C5b-9. Cell Death Dis. 2018, 9, 150. [Google Scholar] [CrossRef]
- Ozen, A.; Kasap, N.; Vujkovic-Cvijin, I.; Apps, R.; Cheung, F.; Karakoc-Aydiner, E.; Akkelle, B.; Sari, S.; Tutar, E.; Ozcay, F.; et al. Broadly effective metabolic and immune recovery with C5 inhibition in CHAPLE disease. Nat. Immunol. 2021, 22, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Young, N.S.; Schubert, J.; Brodsky, R.A.; Socié, G.; Muus, P.; Röth, A.; Szer, J.; Elebute, M.O.; Nakamura, R.; et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2006, 355, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- West, E.E.; Woodruff, T.; Fremeaux-Bacchi, V.; Kemper, C. Complement in human disease: Approved and up-and-coming therapeutics. Lancet 2024, 403, 392–405. [Google Scholar] [CrossRef]
- Yang, J.; Ahn, H.N.; Chang, M.; Narasimhan, P.; Chan, P.H.; Song, Y.S. Complement component 3 inhibition by an antioxidant is neuroprotective after cerebral ischemia and reperfusion in mice. J. Neurochem. 2013, 124, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Yoon, N.G.; Lee, J.E.; Hu, S.; Yoon, S.; Kim, S.Y.; Hong, J.H.; Nam, D.; Chae, Y.C.; Park, J.B.; et al. Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Exp. Mol. Med. 2020, 52, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Bahreyni, A.; Mohamud, Y.; Luo, H.L. Emerging nanomedicines for effective breast cancer immunotherapy. J. Nanobiotechnol. 2020, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Luan, X.; Lei, T.; Fang, J.; Liu, X.; Fu, H.; Li, Y.; Chu, W.; Jiang, P.; Tong, C.; Qi, H.; et al. Blockade of C5a receptor unleashes tumor-associated macrophage antitumor response and enhances CXCL9-dependent CD8(+) T cell activity. Mol. Ther. 2024, 32, 469–489. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; Hwang, S.; Luo, H.; Mohamud, Y. Double-Edged Sword: Exploring the Mitochondria–Complement Bidirectional Connection in Cellular Response and Disease. Biology 2024, 13, 431. https://doi.org/10.3390/biology13060431
Lin J, Hwang S, Luo H, Mohamud Y. Double-Edged Sword: Exploring the Mitochondria–Complement Bidirectional Connection in Cellular Response and Disease. Biology. 2024; 13(6):431. https://doi.org/10.3390/biology13060431
Chicago/Turabian StyleLin, Jingfei (Carly), Sinwoo (Wendy) Hwang, Honglin Luo, and Yasir Mohamud. 2024. "Double-Edged Sword: Exploring the Mitochondria–Complement Bidirectional Connection in Cellular Response and Disease" Biology 13, no. 6: 431. https://doi.org/10.3390/biology13060431
APA StyleLin, J., Hwang, S., Luo, H., & Mohamud, Y. (2024). Double-Edged Sword: Exploring the Mitochondria–Complement Bidirectional Connection in Cellular Response and Disease. Biology, 13(6), 431. https://doi.org/10.3390/biology13060431